M. Bower; S. Collins; C. Cottrill; K. Cwynarski; S. Montoto; M. Nelson; N. Nwokolo; T. Powles; J. Stebbing; N. Wales; A. Webb
HIV Med. 2008;9(6):336-388. ©2008 Blackwell Publishing
Posted 12/15/2008
HIV infection is associated with three AIDS-defining malignancies (Kaposi’s sarcoma, high-grade B-cell non-Hodgkin’s lymphoma and invasive cervical cancer) as well as an increased risk of a number of other malignancies. The clinical care of patients with these tumours requires a multidisciplinary approach drawing on the skills and experience of all healthcare professional groups. Moreover, optimal care can only be achieved by the close co-operation of oncologists, haematologists and HIV physicians, and unless all these clinicians are intimately involved in the care of patients it is likely that the outcome will be less favourable. Patients with HIV-associated malignancies should therefore only be managed in a centre dealing with large numbers of patients with these tumours. An audit study in North London confirmed the better management of patients with AIDS-related lymphoma in HIV centres with cohorts of >500 patients (level of evidence IV C).[1] We recommend that all patients with HIV and malignancy should be referred to centres that have developed expertise in the management of these diseases and serve an HIV cohort of >500. The multidisciplinary medical team managing these patients must include HIV physicians, oncologists, haematologists and palliative care physicians. In line with national cancer waiting times, all patients with suspected cancers must be referred urgently and seen within 2 weeks of referral. Moreover, the NHS Cancer Plan sets out the goal that no patient should wait longer than 1 month from an urgent referral with suspected cancer to the start of treatment.
The early chapters of these guidelines consider the three AIDS-defining malignancies, Kaposi’s sarcoma, high-grade non-Hodgkin’s lymphoma (including primary cerebral lymphoma) and cervical cancer. These chapters are followed by chapters on the non-AIDS-defining malignancies including anal cancer, Hodgkin’s lymphoma, multicentric Castleman’s disease and other non-AIDS-defining malignancies, whilst the final chapter discusses the role of antiretroviral therapy and opportunistic infection prophylaxis in the management of malignancy in people with HIV infection.
These guidelines have used the British HIV Association (BHIVA) standard grading for levels of evidence (see Table 1 ).
Reference
Brook MG, Jones K, Bower M, Miller RF. Management of HIV-related lymphoma in HIV treatment centres in North Thames Region. Int J STD AIDS 2004; 15: 765-766.
2.0 Kaposi’s Sarcoma
2.1 Diagnosis, Staging and Prognosis
Kaposi’s sarcoma (KS) is the most common tumour in people with HIV infection and is an AIDS-defining illness. The cutaneous lesions are characteristic and often diagnosed clinically. The diagnosis can be confirmed histologically and graded into patch, plaque or nodular grade disease. Visceral disease is uncommon, affecting about 10% at diagnosis, and computed tomography (CT) scans, bronchoscopy and endoscopy are not warranted in the absence of symptoms.
The AIDS Clinical Trial Group (ACTG) staging system for AIDS-related KS was developed in the pre-highly active antiretroviral therapy (HAART) era to predict survival and unlike most cancer staging schemes includes tumour-related criteria (T), host immunological status (I) and the presence of systemic illness (S) (see Table 2 ).[1,2] The ACTG also established uniform criteria for response evaluation in AIDS KS (see Table 3 ).[1] In the era of HAART the prognostic value of this staging system has been questioned and one study suggested that only the T and S stages identified patients with a poor survival prognosis.[3] However, a comprehensive evaluation of prognostic factors in 326 patients diagnosed with AIDS-KS in the era of HAART, externally validated on 446 patients from the US HIV/AIDS Cancer Match Study, has established a prognostic score.[4] Having KS as the first AIDS-defining illness (-3 points) and increasing CD4 cell count (-1 for each complete 100 cells/µL in counts at KS diagnosis) improved prognosis, whereas age at KS >50 years (+ 2) and S1 stage (+ 3) conveyed a poorer prognosis. On the basis of this index it was suggested that patients with a poor risk prognostic index (score >12) should be initially treated with HAART and systemic chemotherapy together whilst those with a good risk prognostic index (score <5) should be treated initially with HAART alone, even if they have T1 disease.
2.2 Management
2.2.1 Prevention. The introduction of HAART was associated with a substantial reduction in the incidence of KS in many large cohorts.[5-9] Some of this decline in incidence appears to have preceded the introduction of HAART.[10] However, cohort studies have demonstrated that HAART protects against the development of KS and that nonnucleoside reverse transcriptase inhibitor (NNRTI)-based regimens are as effective as protease inhibitor (PI)-based regimens in terms of their protection.[6] In contrast, the incidence of KS continues to rise in Africa.[11-14]
Specific therapies against human herpesvirus-8, the cause of KS, may also be helpful although these are unlikely to be effective against established lesions which contain mainly latent rather than lytic virus. A UK cohort study of 3688 people with HIV showed that the risk of KS was reduced by ganciclovir and foscarnet exposure but not acyclovir.[15] However, data from a cohort of 935 homosexual men with AIDS found that neither acyclovir, nor ganciclovir, nor foscarnet significantly reduced the risk of KS.[16]
2.2.2 Treatment. 2.2.2.1 Local Therapy. Local treatments are most useful for managing localized bulky KS lesions or for cosmesis. However, local therapies are limited by their inability to affect the development of new lesions in untreated areas.
2.2.2.2 Radiotherapy. During the pre-HAART era radiotherapy had an important and established role in the management of low-volume cutaneous KS, including the cosmetic control of skin lesions and the treatment of painful lesions on the soles, on the genitalia, in the oral cavity and on the conjunctiva.[17] An early randomized study of radiation fractionation for cutaneous KS showed that both response rate and duration of local control were better with fractionated regimens (40 Gy in 20 fractions and 20 Gy in 10 fractions) compared with an 8 Gy single fraction, although toxicity and patient convenience were worse.[18] A second nonrandomized study of 57 patients found no significant difference in response rates between 16 Gy in four fractions and 8 Gy in a single fraction.[19] A retrospective study of 80 patients including some with endemic KS treated with a radiotherapy dose of 8 Gy reported an objective response rate of 74%.[20] In another study of 36 patients with KS of the feet, with a schedule of 3 fractions/week at 3.5 Gy/fraction, up to a total dose of 21 Gy, the response rate was 91% with a complete response rate of 80%.[21]
However, the side effects of radiotherapy in people with AIDS are often severe.[17,22] This is particularly notable in the oral cavity and on the soles of the feet. Modified fractionated schedules and close attention to skin care including avoidance of friction and sparing use of moisturisers are required to keep toxicity as low as possible. The explanation for this increased toxicity is not clear. Although the use of radiotherapy in the management of KS has declined since the introduction of HAART, it still maintains an important role in the management of KS at specific sites. 90Strontium brachytherapy is an effective and well-tolerated treatment for eyelid and conjunctival lesions.[23]
An important large randomized study from Zimbabwe has evaluated treatments for AIDS-KS in 495 patients who were not treated with antiretroviral (ARV) agents. This showed that radiotherapy did not improve either overall survival or quality of life compared with supportive care alone.[24] Although discomfort from radiotherapy is frequent, it usually resolves without intervention within 2 weeks of completion of therapy.
2.2.2.3 Other Local Therapies. Alitretinoin gel (0.1%) (9-cis-retinoic acid) is a topical, self-administered therapy approved for the treatment of KS in the USA but not licensed in Europe. Two double-blind, randomized placebo-controlled trials, involving a total of 402 individuals, evaluated 12 weeks of twice-daily alitretinoin gel.[25,26] The response rates in the active arm after 12 weeks were 37%[26] and 35%[25] compared with 7 and 18% in the placebo arms analysed by intention to treat. In both studies over 80% of participants were receiving HAART and this did not influence the results. The gel may cause dermal irritation and some undesirable skin lightening at the application site. Responses are typically noted in patients with a wide variety of CD4 cell counts and typically occur 4-8 weeks after treatment.
Local problems such as gastrointestinal bleeding, perforation, volvulus and intussusception may be treated surgically, but surgery including amputation is no longer indicated in the routine management of this disease. Intralesional vinblastine is the most widely used intralesional agent and responses of around 70% were reported in the pre-HAART era.[27,28] Treated lesions usually fade and regress although typically do not resolve completely. A randomized study in 16 patients comparing intralesional vinblastine or sodium tetradecyl sulphate in the treatment of oral KS demonstrated partial responses in both groups with no significant differences.[29] Intralesional injections of biological agents such as interferon (IFN)-α have also shown activity, but are infrequently used now.
In one early study of 20 patients, complete responses were observed in 80% of lesions treated with cryotherapy, and the duration of the response was more than 6 weeks. Greater than 50% cosmetic improvement of KS was reported in this pre-HAART era study.[30] An alternative experimental approach is photodynamic therapy, which is based upon activation by light of a photosensitizing drug that preferentially accumulates in tumour tissues such as KS.[31] A series of 25 patients with a total of 348 KS lesions received photofrin 48 h prior to light activation. No patients were on HAART and 95% of the lesions responded to therapy (33 and 63% complete and partial responses, respectively).[32] To a large extent local therapies for KS have been superseded by the introduction of HAART.
2.2.2.4 Systemic Therapy. 2.2.2.4.1 HAART. There are no randomized trials comparing HAART with no HAART as all patients with KS should receive HAART. Many case reports and small series have described the regression of KS with HAART (and individual ARVs), and HAART, alone and in combination with other treatment modalities (local therapy, systemic therapy, immunotherapy, biological therapy and radiotherapy), has been shown to prolong time to treatment failure in KS[33] and to prolong survival in patients who have been treated for KS with chemotherapy.[34] No difference has been demonstrated in time to progression between patients receiving a PI-based HAART regimen and those receiving an NNRTI-based HAART regimen despite the anti-angiogenic effects of PIs observed in the laboratory.[33]
The effect of HAART on KS is highlighted by the Swiss cohort study: the relative risk of KS development between 1997 and 1998 (HAART era) compared with the time period between 1992 and 1994 (pre-HAART era) was 0.08 [95% confidence interval (CI) 0.03-0.22].[35] A further European cohort study reported a significant reduction in the cumulative probability of KS on HAART.[36]
2.2.2.4.2 Cytotoxic chemotherapy. Administration of systemic cytotoxic chemotherapy is warranted in patients with more advanced or rapidly progressive disease. It has been suggested that patients with a poor risk prognostic index (score > 12) should be initially treated with HAART and systemic chemotherapy together whilst those with a good risk prognostic index (score < 5) should be treated initially with HAART alone, even if they have T1 disease.[4] However, the decision to initiate systemic chemotherapy is usually based on a number of parameters including the prognostic index, response to HAART alone, patient performance status and end organ function, including hepatic and bone marrow reserve. Typical indications for systemic chemotherapy include widespread skin involvement such as more than 20 lesions, extensive KS of the oral cavity, tumour-associated oedema or ulceration, symptomatic visceral involvement and immune reconstitution inflammatory syndrome-induced KS flare.[37] In the pre-HAART era, several chemotherapeutic agents (bleomycin, doxorubicin, vinblastine, vincristine and etoposide) were shown to have activity against KS in case series and small phase II trials using different combinations and doses of these drugs.[38-42] However, liposomal anthracyclines and taxanes have become established as the backbone of current standard systemic cytotoxic therapy against KS.
2.2.2.4.3 Liposomal anthracyclines. Liposome encapsulation of anthracyclines constitutes a considerable advance in the chemotherapy of KS. The advantages of liposomal formulation include increased tumour uptake and hence favourable pharmacokinetics. The trials of liposomal anthracyclines for HIV-associated KS were undertaken in the pre-HAART era but clinicians continue to regard them as the gold-standard first-line chemotherapy for KS. Both liposome-encapsulated daunorubicin (DaunoXome 40 mg/m2 every 2 weeks) and the pegylated liposomal doxorubicin, which is known variously as Caelyx, Doxil or PLD (20 mg/m2 every 3 weeks), have been shown to have good antitumour activity. The toxicity profile is better than for other anthracyclines, with no reported cardiotoxicity even at high cumulative dosages[43] and rarely significant alopecia; however, there remains considerable myelosuppression, and occasional emesis. In addition, infusion-related hypotension and hand/foot syndrome are novel side effects seen with these liposomal formulations.
Three sizeable, randomized controlled studies have compared liposomal anthracyclines with conventional combination chemotherapy regimens and all were conducted before the introduction of HAART. A phase III randomized comparison of DaunoXome and ABV (doxorubicin, bleomycin and vincristine) demonstrated equivalent overall response rates (partial and complete responses), time to treatment failure and survival duration.[44] Two randomized phase III trials compared pegylated liposomal doxorubicin with conventional combination chemotherapy [ABV in one study and BV (bleomycin and vincristine) in the other], as first-line therapy for KS in patients not on HAART. Both found response rates were higher in the Doxil arms but responses were often not sustained[45,46] (see Table 4 for details). The three phase III studies may not be directly comparable. In one small randomized study in 79 patients, KS patients were randomized to PLD (20 mg/m2) or DaunoXome (DNX) (40 mg/m2) every 2 weeks for up to six cycles; nonsignificant differences favouring PLD were found, although the study was under-powered and there is insufficient evidence for a recommendation of which liposomal anthracycline to use.[47]
Since the widespread introduction of HAART, the duration of responses to treatment for KS have increased[48] and no further randomized trials have compared liposomal anthracyclines with nonencapsulated chemotherapy regimens. The safety and tolerability of these drugs in combination with HAART have been evaluated. In one study of 54 patients, 82% had a response within 8 weeks and the PLD/HAART combination was well tolerated with no evidence of suppression of CD4 cell counts.[49] In a cohort study of 50 patients treated with concomitant HAART and liposomal anthracycline chemotherapy for KS, there was no decline in CD4 cell count or rise in HIV viral load.[50] These findings suggest that standard opportunistic infection prophylaxis guidelines may be followed when treating patients with liposomal anthracycline chemotherapy for KS. Based on the response rates, median response durations and the toxicity profile, liposomal anthracyclines are considered first-line chemotherapy for advanced KS.
2.2.2.4.4 Taxanes. The major mechanism of cytotoxicity of taxanes, including paclitaxel, which is approved for KS treatment, is attributed to binding to β-tubulin polymers, which stabilizes microtubules against depolymerization. Paclitaxel also promotes apoptosis and down-regulates Bcl-2 protein expression in KS cells in vitro and in KS-like lesions in mice.[51,52]
In a number of phase II trials, paclitaxel was shown to have single-agent activity against AIDS-KS; furthermore, these studies included a number of patients who had previously received anthracyclines.[53-57] In one phase II study of paclitaxel (135 mg/m2 every 3 weeks) for KS, 28 patients were enrolled and a response rate of 71% was reported. As a whole, this included four patients (14%) who had received anthracyclines but no patients received HAART.[54] A second, larger study of 56 patients included 20 patients (36%) who received a PI at some stage during the study and 40 (70%) who had received prior therapy for KS, which included liposomal anthracyclines in 17 patients (30%). The overall objective response rate was 59% (amended ACTG criteria), and the median response duration was 10.4 months.[55]
Subsequently two studies have addressed the role of paclitaxel as second-line chemotherapy. In one open-label multicentre trial, 107 individuals were enrolled who had received prior chemotherapy for AIDS-KS. The previous therapy regimens included ABV (adriamycin, bleomycin and vincristine) in 52 patients, liposomal daunorubicin in 49 patients, and liposomal doxorubicin in 40 patients. Moreover, only 77% were receiving concomitant HAART (all PI-based) and 33% started this treatment at the same time as the taxane chemotherapy. The paclitaxel protocol used was 100 mg/m2 fortnightly. The overall response rate was 56% with no significant difference in response rate when comparing patients on or not on HAART. Less surprising was the finding that patients on HAART had a significantly improved survival. The main side effect reported in these studies was neutropenia – this generally resolved prior to the next cycle.[56]
In a second study of 17 patients with anthracycline-refractory AIDS-KS that had progressed during or within 6 months of completing liposomal anthracycline chemotherapy, all patients were receiving a stable HAART regimen to avoid confounding of results. The treatment schedule was again 100 mg/m2 fortnightly. The objective response rate to paclitaxel was 71% (95% CI 60-81); eight (of 17) partial responses and four (of 17) complete responses. There were no significant changes in CD4, CD8, CD16/56 (natural killer cells) and CD19 (B cells) lymphocyte subset cell counts during and for up to 1 year following chemotherapy. Similarly, plasma HIV-1 viral loads did not change significantly during or after treatment, suggesting that the combined use of paclitaxel and HAART reduces the risk of chemotherapy-related immunological decline and opportunistic infections.[58] In contrast, previous trials without concomitant HAART were worrying in this respect; Gill[55] reported 51 AIDS-defining opportunistic infections in the 56 patients treated with paclitaxel (10.5/100 patient-months on paclitaxel), only 36% of whom received HAART, and Welles et al. reported 27 opportunistic infections (8.4/100 person-months on paclitaxel) among their cohort of 28, none of whom received HAART.[54] Thus the concomitant use of HAART and paclitaxel appears to be safe and not detrimental to immune function despite initial concerns about pharmacological interactions.[59] These findings suggest that standard opportunistic infection prophylaxis guidelines may be followed when treating patients with taxane chemotherapy for KS.
The higher prevalence rates of alopecia, myalgias and myelosuppression and the need for a 3-h infusion make paclitaxel a less attractive first-line option than PLD. Moreover, the need for corticosteroid administration (typically dexamethasone 10-20 mg intravenously 30 min prior to paclitaxel, or 10 mg orally 12 and 6 h prior) to prevent allergic reactions raises further concerns for some clinicians.
The clinical experience with docetaxel in KS is much more limited, although two small studies suggest that this agent can produce meaningful responses when used weekly,[60] or in anthracycline pretreated individuals.[61]
2.2.2.4.5 Immunotherapy. The biological response modifier IFN-α was approved for KS treatment before the availability of HAART and liposomal anthracyclines.
The ACTG randomized 68 individuals to low- and intermediate-dose IFN-α (1 million and 10 million units daily, respectively) plus didanosine.[62] Response rates and durations were not statistically different although there were more toxicities in the higher dose group. In another randomized study, 108 patients were treated with IFN-α (1 million or 8 million units daily) with zidovudine.[63] The higher dose regimen was associated with a statistically higher response rate and longer time to progression. In a retrospective study of patients with classic KS comparing PLD with low-dose IFN-α, 12 patients received 20 mg/m3 of PLD monthly while six received 3 million units of IFN-α three times per week, and PLD was found to be superior in terms of responses and toxicity.[64]
Response to IFN-α frequently requires continued treatment for 6 months or more, as the time to response is typically more than 4 months. It should not be considered for progressive or visceral disease. Toxicity at higher doses including fever, chills, neutropenia and depression is common, and poor responses are observed in the setting of low CD4 cell counts. While it can be considered in those with residual KS who have appropriately reconstituted their immune systems with HAART, it is seldom used.
2.2.2.5 Other Systemic Therapies. Thalidomide has significant anti-angiogenic activity. A total of 37 patients were enrolled in two phase II studies. Partial responses were recorded for 35 and 47% of evaluable patients, with toxicity including fatigue, neuropathy and depression.[65,66] The importance of the c-kit pathway has been evaluated in 10 patients with previously treated cutaneous KS who received oral imatinib; half achieved a partial response but diarrhoea necessitated dose reduction in 60%.[67] Other therapies are being developed including COL-3, a matrix metalloproteinase inhibitor which in a phase II trial of 75 patients demonstrated partial responses in 41%.[68] Similarly, interleukin (IL)-12 was administered to patients on HAART with KS and the response rate was 71%.[69] Unlike other therapies discussed above, neither COL-3 nor IL-12 has been approved for use in any disease.
A number of anti-herpes virus agents have been studied in AIDS-related KS; none has demonstrated significant activity, although they have been shown to prevent KS in one cohort study.[15]
A Cochrane Database systematic review which focused on five trials involving 915 individuals concluded that alitretinoin gel or radiotherapy is effective in treating cutaneous KS and PLD is effective treatment for advanced KS. Interestingly, this systematic review found that only radiotherapeutic options were applicable to resource-poor settings.[70]
2.3 Summary of Recommendations
Early-stage KS (T0 stage)
|
Advanced-stage KS (T1 stage)
|
2.4 References
|
3.0 Systemic AIDS-related Non-Hodgkin’s Lymphoma
3.1 Introduction
HIV-infected patients are at an increased risk of developing non-Hodgkin’s lymphoma (NHL),[1-3] and AIDS-related non-Hodgkin’s lymphoma (ARL) is an AIDS-defining illness (ADI). This is the second most common tumour in individuals with HIV and, although studies show a decline in incidence in the HAART era,[4-6] ARLs have increased as a percentage of first ADI.[7,8]
The development of ARL has been shown to be related to older age, low CD4 cell count and no prior treatment with HAART.[9] Patients tend to present with advanced-stage disease, B symptoms, extranodal sites of disease and bone marrow involvement. The incidence of central nervous system (CNS) involvement is higher in ARL compared with HIV-negative patients with NHL.[10,11]
Before the introduction of HAART, the outlook for patients with ARL was poor, with the median survival time for patients treated with chemotherapy being around 2-13 months. Median survival in the post-HAART era is now nearing 24 months[12,13] and is beginning to approach that observed in the HIV-negative population, and depends critically on histological subtype and stage of disease.[14]
3.2 Diagnosis, Staging and Prognosis
The diagnosis of ARL should be based on a tissue sample rather than a cytological sample. In addition to the routine investigations advised as part of HIV clinical care, all patients require staging with clinical evaluation, blood tests, CT scanning and bone marrow aspiration and trephine (see Table 5 ).
All patients should have pathology and treatment plans reviewed by a specialist multidisciplinary team (MDT). Staging should be according to the Ann Arbor classification/Cotswolds modification system (see Table 6 ).
Prognostic factors for survival in the pre-HAART era were predominantly immunological (prior ADI and low CD4 cell count).[15,16] Factors that are associated with decreasing survival in the post-HAART era are increasing International Prognostic Index (IPI) scores and failure to attain complete remission on completion of chemotherapy[17,18] particularly the latter in one retrospective study, although response to therapy is of course not available at diagnosis.[19] A prognostic index in which a CD4 cell count is added to the widely used IPI (based on age, stage, serum lactate dehydrogenase (LDH), performance status and number of extranodal disease sites; see Table 7 [20]) has been established in ARL.[21] Weighting scores are given to a high IPI score (2.9), a high-intermediate IPI score (1.84), and a CD4 count <100 cells/µL (1.34). The prognostic risk scores are divided into quartiles: <1.0, 1.0 to 1.83, 1.84 to 2.90, and >2.90. These validated risk strata can predict 1-year survival rates of 82, 47, 20 and 15%.
3.3 Management
3.3.1 Diffuse Large B-cell Lymphoma. 3.3.1.1 First-line Chemotherapy for Diffuse Large B-cell Lymphoma in HIV-infected Individuals. Prior to the introduction of HAART, treatment with standard dose chemotherapy induced high levels of toxicity and a high incidence of opportunistic infections. The introduction of haemopoietic growth factors into treatment protocols has allowed the introduction of increasingly myelotoxic regimens. Prior to the introduction of HAART, improvements in chemotherapy response rates were generally offset by increased numbers of deaths as a result of opportunistic infection. Clinical trials were conducted in the pre- and post-HAART eras to investigate the possibility of effective treatment with reduced-dose chemotherapy.
In a multicentre trial in the pre-HAART era, 192 patients were randomized to either full-dose MBACOD (methotrexate, bleomycin, doxorubicin, cyclophosphamide, vincristine and dexamethasone) chemotherapy with granulocyte-macrophage colony-stimulating factor (GM-CSF) support or low-dose MBACOD in ACTG-142.[22] There were no significant differences in median survival (7.7 months for the low dose; 6.8 months for the standard dose) or response rate (41% for the low dose; 52% for the standard dose). There was an increased incidence of grade 4 neutropenia in those receiving standard-dose chemotherapy (69% for the full dose vs. 50% for the low dose; P>0.007) but this did not result in a significant difference in rates of febrile neutropenia. There was, however, an increased rate of lymphoma-associated death in the low-dose chemotherapy arm.
The European Intergroup conducted a randomized control trial predominantly in the pre-HAART era; 485 patients were randomly assigned to chemotherapy after risk stratification according to a three-point HIV score based on performance status (1 point), prior AIDS (1 point), and CD4 cell count below 100 cells/µL (1 point). Two hundred and eighteen good-risk patients (HIV score 0) received ACVBP (doxorubicin, cyclophosphamide, vindesine, bleomycin and prednisolone) or CHOP (doxorubicin, cyclophosphamide, vincristine and prednisolone); 177 intermediate-risk patients (HIV score 1) received CHOP or low-dose CHOP (Ld-CHOP); and 90 poor-risk patients (HIV score 2-3) received Ld-CHOP or VS (vincristine and steroid). Response rates and survival times were not significantly different between the treatment arms in each group; the only significant differences were for use of HAART, HIV score and IPI, not chemotherapy regimen.[23] The only statistically significant difference between treatment arms was a higher response rate in the CHOP arm compared with the Ld-CHOP arm (CR/CRu of 49 vs. 32%; P = 0.02) in the patients designated intermediate-risk (HIV score 1).
These results are consistent with those from randomized trials of chemotherapy in the HIV-negative population for diffuse large B-cell lymphoma (DLBCL), in which CHOP is considered the standard therapy for the majority of patients. Prior to the introduction of rituximab, no survival advantage over CHOP was demonstrated for any other chemotherapy regimens.[24-26] Increasingly, a treatment strategy resembling that for the management of aggressive NHL in the immunocompetent population has been accepted for patients with HIV infection, and so we discuss the management of DLBCL, and Burkitt’s (BL) or Burkitt-like lymphomas, separately.
3.3.1.2 The Effect of Adding HAART. A comparison of 363 patients treated before and after the introduction of HAART has shown that overall survival has improved in the HAART era.[27] Although tumour regressions with immune reconstitution are not observed with lymphomas, optimizing the immune status of the patient has been shown to reduce opportunistic infections and is associated with superior response rates and survival.[28-31]
The German ARL study group investigated whether HAART administered concomitantly with CHOP improved outcomes. They used an adjusted IPI in 72 individuals and found that concurrent CHOP plus HAART was safe and effective, with no adverse effects on CD4 cell count.[32] Case-control series have compared treatment with CHOP in the pre- and post-HAART eras and have reported higher response rates and improved survival with the addition of HAART to CHOP chemotherapy.[29,30] Other phase II studies using CHOP and HAART therapy have reported complete remission (CR) rates of between 48 and 92% and median survival times of between 15 and >34 months.[28]
The AIDS Malignancy Consortium investigated the efficacy and toxicity of combining low- or standard-dose CHOP chemotherapy with HAART.[33] Forty patients received reduced doses of cyclophosphamide, doxorubicin, vincristine and prednisolone (modified CHOP), and 25 subsequent patients received full-dose CHOP with granulocyte colony-stimulating factor (G-CSF). The CR rates were significantly higher in the full-dose CHOP arm (48%, as compared with 30% in the modified CHOP arm). No long-term outcome data have been reported for this group of patients, but treatment-related toxicity was similar between the two groups.
There are concerns that HAART may interact with chemotherapy and cause adverse drug reactions that may limit the chance of cure. The National Cancer Institute developed a dose-adjusted schedule for EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide and doxorubicin) chemotherapy in which HAART was omitted during chemotherapy. Despite a high response rate, CD4 cell counts fell dramatically during chemotherapy and took 12 months to recover to baseline levels despite the re-introduction of HAART on completion of chemotherapy.[34]
3.3.1.3 Infusional Chemotherapy for Diffuse Large B-cell Lymphoma. The activity of infusional chemotherapy regimens for the treatment of non-HIV-associated lymphoma was first reported in 1993 using the combination of cyclophosphamide, doxorubicin and etoposide (CDE) administered as a 96-h continuous infusion for up to six courses at 4-weekly intervals together with G-CSF.[35] In a selected group of 25 patients with ARL who were treated with infusional CDE and didanosine, the median survival time was 18.4 months.[36] The same schedule was then combined with the PI saquinavir, which produced similar results but with an increased incidence of treatment-associated mucositis.[37]
Overall the single institution experience in 62 patients is an observed CR rate of 53% and a median survival time of 18 months. The Eastern Cooperative Oncology Group conducted a multicentre phase II trial of infusional CDE in 98 patients with ARL during the period of HAART introduction. The overall results showed a CR rate of 45% and a median survival time of 12.8 months. However, patients who received HAART did better, with a median survival time of 13.8 months compared with 6.8 months for those not receiving HAART.[38]
3.3.1.4 Rituximab for Diffuse Large B-cell Lymphoma. Rituximab is a monoclonal humanized antibody that targets CD20 on the surface of B cells. The addition of rituximab to CHOP has been shown to prolong event-free and overall survival in HIV-negative patients with DLBCL.[39] The benefit of rituximab is most evident in BCL6-negative cases which are mostly of the activated B-cell (ABC) type.[40] CHOP-R is considered standard therapy for HIV-negative patients with DLBCL in the UK. Currently, a national randomized trial is comparing the outcomes of patients receiving CHOP-R on a 14- or 21-day treatment cycle.
A number of prospective studies have addressed the impact of rituximab in HIV-positive individuals with DLBCL. The first of these, a randomized trial in 149 patients, compared CHOP-R (n = 99) with CHOP (n = 50), using a standard rituximab dose of 375 mg/m2 with each cycle of chemotherapy and 3-monthly maintenance doses of rituximab in complete or partial responders to R-CHOP.[41] The use of rituximab was associated with a significant reduction in the progression of lymphoma on treatment, and in death caused by lymphoma.[41] However, an increase in infectious deaths was observed in rituximab-treated patients, especially (nine of 15) in patients with CD4 cell counts <50 cells/µL. In this study, six of 15 deaths occurred during the maintenance phase of rituximab treatment, a strategy not routinely used in aggressive NHL in HIV-negative patients. An increased risk of life-threatening infection was also observed when three phase II studies were pooled, combining rituximab with the infusional CDE chemotherapy regimen in 74 patients with ARL.[42] However, in another phase II trial involving 61 patients with ARL, rituximab adjunction to CHOP has been shown to be efficacious without increasing the risk of life-threatening infections.[14] However, in this trial inclusion criteria precluded advanced HIV infection, and only four patients had CD4 cell counts <50 cells/µL. Presently, caution in the use of rituximab, especially in patients with CD4 cell counts <50 cells/µL, is advocated. This area remains contentious and results from further trials are awaited.
3.3.1.5 Second-line Therapy for Systemic AIDS-related Non-Hodgkin’s Lymphoma. Treatment of refractory or relapsed ARL in the pre-HAART era was disappointing, with few clinically useful responses.[43-45] Since the introduction of concomitant HAART therapy a number of studies have presented more optimistic results. A phase II study reported encouraging results with the salvage regimen ESHAP (etoposide, methylprednisolone, high-dose cytarabine and cisplatin) with a response rate of 54%, although the haematological toxicity was considerable in almost all patients.[46,47] However, the median survival time of the 13 patients in this study was only 7 months.
In the HIV-negative setting, studies have shown that high-dose therapy with autologous stem cell transplantation is the optimal therapy for relapsed NHL.[48] Improvements in the immune function and haematological reserves of HAART-treated patients, and better supportive care, have made stem cell mobilization and high-dose chemotherapy approaches possible in HIV-infected patients. A number of case reports and small series (n = 10-20) have described successful autologous stem cell transplantation in relapsed/refractory patients with ARL.[49-58] Krishnan et al. reported their experience of autografting 20 relapsed/refractory patients with chemosensitive ARL.[55] Stem cell mobilization and engraftment were comparable to those in non-HIV-infected patients. Toxicity was appreciable but manageable. Although opportunistic infections were observed in six patients, all responded to therapy. Their results were impressive, with a progression-free survival of 85% (95% CI 69-100), and overall survival of 85% with a median follow-up period of 31.8 months. Similarly encouraging results have been reported in other series.[54,56-58] These reports suggest that suitable patients with chemosensitive relapsed ARL should now be considered for high-dose chemotherapy and haematopoietic stem cell transplantation.
3.3.1.6 Recommendations for Diffuse Large B-cell Lymphoma.
|
3.3.2 Burkitt’s Lymphoma. Until recently, patients with HIV-associated Burkitt’s lymphoma (BL) have been treated similarly to HIV-positive patients with DLBCL. However, in a large retrospective study the survival of patients with BL was very poor compared with patients with DLBCL, despite adjunctive HAART, if similarly treated with CHOP or MBACOD. The authors suggested that more intensive regimens should be considered for these patients.[27] This suggestion was corroborated by the results of a phase II prospective study involving 74 patients with HIV-NHL and HIV-BL treated with rituximab and the CDE infusional regimen (R-CDE). In multivariate analysis, a diagnosis of HIV-BL was significantly associated with a worse outcome in comparison to HIV-NHL patients.[42] Two small retrospective comparative studies have demonstrated the feasibility of administering more intensive chemotherapy regimens, as used for HIV-negative BL patients, such as CODOX-M/IVAC (cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide and cytarabine)[59] and hyperCVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone, methotrexate and cytarabine).[60] In these studies, toxicity and outcome were similar to those observed in non-HIV-infected patients with BL treated with the same regimen.[60] The 2-year event-free survival was significantly better in BL patients treated with CODOX-M/IVAC compared with those receiving less intensive chemotherapy.[59] These studies, although small and retrospective, suggest that the approach of using a uniform regimen for all pathological subtypes of aggressive ARL should be re-evaluated.
3.3.2.1 Recommendations for Burkitt’s lymphoma.
|
3.3.3 Leptomeningeal Lymphoma Management: Prophylaxis and Treatment. Involvement of the CNS in ARL is associated with a poor prognosis and tends to occur in advanced disease.[61] Secondary spread to the CNS may occur either at presentation of ARL or as a site of disease relapse. The latter is thought to occur because the CNS may be a pharmacologically privileged compartment that is protected from the effects of intravenously administered cytotoxic chemotherapy.
The identification of patients at risk of CNS relapse remains inconclusive;[10] however, data from immunocompetent individuals suggest that BL, advanced stage, young age, elevated serum LDH and B symptoms, along with extranodal disease sites such as testes, paranasal sinuses, paraspinal disease and bone marrow, predict a higher likelihood of CNS relapse. Thus ARL patients with any of these characteristics, as well as those with paraspinal or paranasal disease, should be offered intrathecal prophylaxis. Eleven studies[28-30,33,36,62-67] have reported the use of CNS prophylaxis and treatment in individuals with ARL. Only two were prospective or randomized,[22,41] and these trials allowed individual institutions to administer CSF prophylaxis according to local protocol or preference. Both intrathecal methotrexate (10-15 mg) and intrathecal cytarabine (40-50 mg) were used to prevent and treat CNS disease and, depending on the perceived risk of CNS relapse, one to six administrations were offered. In the majority of cases, patients received at least one intrathecal instillation of chemotherapy and more aggressive intrathecal administration occurred in those individuals with BL. In general, most centres follow the same protocols that they employ in immunocompetent patients, with all BL patients and selected patients with DLBCL, based on agreed criteria that usually include raised serum LDH and extranodal sites of lymphoma, receiving intrathecal prophylaxis.
Treatment of CNS spread of systemic lymphoma involves whole-brain radiotherapy (total dose 24 Gy) and frequent intrathecal cytarabine and/or methotrexate until CSF cytology is negative. The use of liposomal extended release cytarabine (DepoCyte) in individuals with ARL has been investigated in a small retrospective study of those with ARL meningeal involvement at presentation [defined as abnormal enhancement on a brain CT or magnetic resonance imaging (MRI) scan or lymphoma cells in the CSF]. This study compared survival and CSF protein in patients treated with DepoCyte compared to standard therapy (alternating weekly methotrexate and cytarabine twice a week for 4 weeks, then once a week for 4 weeks, then once every 2 weeks for 8 weeks) in previously treated patients.[68] Efficacy parameters remained unchanged while half the number of intrathecal administrations were required.
3.3.3.1 Recommendations for Meningeal Lymphoma Management.
|
3.3.4 Response Evaluation and Follow-up. Specific response criteria for NHL in HIV-infected patients have not been described, but the International Working Group response criteria defined for the general population are generally used.[69] Thus, assessment after treatment should include whole-body CT scans and a bone marrow biopsy (if the CT scan shows complete response and the bone marrow was involved at diagnosis). Patients with a residual mass should have a positron emission tomography (PET) scan. These investigations should be performed at least 4-6 weeks after the last cycle of chemotherapy.
Regarding follow-up, it is recommended for patients in complete remission after treatment to have an oncology appointment every 3 months for the first and second years, every 6 months for the following 3 years and then annually. Investigations at follow-up should include medical history, physical examination and blood tests. No further surveillance investigations are recommended for patients in complete remission. The very small group of patients who have received radiotherapy should have thyroid function tests regularly and an annual chest X Ray (CXR) if they have had mediastinal radiotherapy (RT), and female patients treated with Mantle RT should have breast surveillance (mammographies/MRI).
3.4 References
|
4.0 Primary Central Nervous System Lymphoma
4.1 Introduction
Primary central nervous system lymphoma (PCL) is defined as a non-Hodgkin’s lymphoma (NHL) confined to the cranio-spinal axis without systemic involvement. It is uncommon in immunocompetent patients but occurs more frequently in patients with both congenital and acquired immunodeficiency. Registry linkage studies confirm the markedly increased relative risk of PCL amongst individuals living with AIDS, with an incidence as high as 2-6% in two early reports,[1,2] a likely consequence of the brain representing a reservoir of active viral replication.[3] Shortly after the introduction of highly active antiretroviral therapy (HAART), a decline in the incidence of PCL was recognized by many clinicians and a meta-analysis of 48 000 individuals confirmed this significant decrease [relative risk 0.42; 99% confidence interval (CI) 0.24-0.75].[4] A subsequent study has shown that the incidence of PCL is lower in the HAART era (1.2 cases per 1000 patient-years; 95% CI 0.8-1.9) than in the pre-HAART era (3.0 cases per 1000 patient-years; 95% CI 2.1-4.0; P<0.001), and overall survival is longer (median survival 32 days, range 5-315 vs. 48 days, range 15-1136 days; log rank P = 0.03).[5]
4.2 Diagnosis, Staging and Prognosis
AIDS-related PCL occurs with a similar distribution across transmission risk groups and all ages, and the tumours are characteristically high-grade diffuse large B-cell or immunoblastic NHL.[6] In patients with HIV, computed tomography (CT) scans of PCL may show ring enhancement in as many as half the cases, whilst in immunocompetent patients with PCL the enhancement is almost always homogeneous.[7,8] Similarly, the presence of Epstein-Barr virus (EBV) in tumour cells is a universal feature of HIV-associated PCL but is not found in other PCLs.[9,10] Thus the biology and clinical features of PCL in people with HIV differ from those of PCL in the immunocompetent population and these differences are reflected in the very different clinical management, disease progression and outcome.
The diagnostic algorithm for the management of cerebral mass lesions in HIV-seropositive patients has included a 2-week trial of anti-toxoplasmosis therapy (sulphadiazine 1 g four times a day, pyrimethamine 75 mg once daily). Patients who fail to respond to this therapy are offered further diagnostic procedures: either a brain biopsy or since 1994 a diagnostic lumbar puncture if there are no contraindications. The presence of EBV in AIDS-related PCL led to the development of a polymerase chain reaction (PCR) amplification test that is now routine and can detect EBV DNA in the cerebrospinal fluid (CSF). This has since become established as a diagnostic test in the presence of a cranial space-occupying lesion, with high sensitivity (83-100%) and specificity (>90%).[11-13] The CSF is examined for EBV DNA by PCR as previously described,[14] and a positive brain biopsy or lumbar puncture confirms a diagnosis of PCL, whilst failure of anti-toxoplasma treatment without further diagnostic intervention is classified as a presumptive diagnosis of PCL.
Thorough evaluation to determine the full extent of disease is critical before the initiation of therapy to ensure that the patient receives appropriate therapy. This evaluation includes studies of the CNS and body, and consideration of bone marrow aspirate and trephine. Optimal imaging of the brain parenchyma requires a gadolinium-enhanced magnetic resonance imaging (MRI) scan. Contrast-enhanced CT scans may be substituted in patients in whom MRI is medically contraindicated (e.g. cardiac pacemaker) or unavailable. All patients should have a lumbar puncture for CSF cytology unless medically contraindicated.[15] Occult systemic disease should and can be excluded by staging with CT scans of the chest, abdomen and pelvis.[16,17] Because patients derive no clinical benefit from surgical resection and the deep-seated nature of most lesions increases the risk of surgical complications, stereotactic needle biopsies may be performed if there are doubts about the diagnosis. If there is evidence of ocular or CSF involvement, a vitrectomy or CSF cytology may establish the tissue diagnosis. Combined single-photon emission computed tomography (SPECT) with EBV DNA in CSF has a very high diagnostic accuracy in HIV-positive patients with a cerebral mass lesion. In a study of 31 HIV-positive patients, 13 with PCL and 18 with nontumour disorders, EBV DNA was never detected in patients with nonneoplastic lesions.[18] For PCL diagnosis, hyperactive lesions showed 92% sensitivity and 94% negative predictive value (NPV), whereas positive EBV DNA had 100% specificity and 100% positive predictive value. The presence of increased uptake and/or positive EBV DNA had 100% sensitivity and 100% NPV. Because PCL is extremely likely in patients with hyperactive lesions and positive EBV DNA, brain biopsy can be avoided, and patients can promptly undergo radiotherapy or multimodal therapy. However, in patients showing hypoactive lesions with negative EBV DNA, empiric anti-toxoplasma therapy is indicated. In patients with discordant SPECT/PCR results, brain biopsy is advisable. Moreover, patients considered for aggressive therapy such as high-dose methotrexate should have the diagnosis confirmed by stereotactic biopsy unless contraindicated.
The baseline evaluation of any newly diagnosed patient with PCL should include a comprehensive physical and neurological examination. Particular attention should be paid to examination of peripheral lymph nodes in all patients and the testes in men. In immunocompetent patients with PCL, age and performance status are the two most widely documented prognostic variables and must be recorded in every patient. At this point, there is no standard battery of neuropsychological testing; some recommend baseline and serial scoring of mini mental state examination (MMSE).[17] Baseline laboratory evaluation should include serum lactate dehydrogenase in all patients and determination of adequate hepatic and renal function in those who will receive high-dose methotrexate.
In immunocompetent individuals with PCL, a median survival time of 9 months has been reported with no consistent improvement in the last three decades of the 20th century.[19] However, a recent series of 338 consecutive immunocompetent patients with PCL reported a median survival time of 37 months.[20] By comparison, the prognosis of AIDS-associated PCL is dismal; the median survival time is generally quoted as 2-3 months, although the point from which it is measured (including completion of anti-toxoplasmosis therapy) varies and not all the patients are included in some series,[21-23] while in others only treated patients[24,25] or those with confirmed diagnoses[26] are included. When tumour progression occurs, it is usually confined to the CNS and/or the eye.
4.3 Management
There is no class I evidence for any therapeutic option in AIDS-PCL, apart from the use of HAART as prevention.[5] Whilst PCL is sensitive to both chemotherapy and radiotherapy, the overall response rates and long-term survival are significantly inferior to the results achieved in similar subtypes of extranodal NHL.[17] Since the introduction of HAART the incidence of primary CNS lymphoma (PCNSL) has dramatically decreased.[27] The standard treatment modality for primary cerebral lymphoma in HIV-infected patients has been whole-brain irradiation and HAART. However, the median survival time in a study involving 111 patients treated in this way was just 3 months.[28] There is no evidence for consolidation therapy and steroids are given for symptoms. Although differing antiretrovirals have differing CSF penetrations, all are associated with a reduced risk of PCL.[5] The aims of treatment in this setting have previously been, therefore, to relieve symptoms, and improve quality of life with minimal adverse effects.[23] Single-agent chemotherapy with intravenous high-dose methotrexate and folinic acid rescue has been studied in AIDS patients with primary cerebral lymphoma in the context of a prospective uncontrolled study that included 15 patients. The results showed a complete response in 47% of patients, a median survival time of 19 months, a low relapse rate of approximately 14% and no evidence of neurological impairment or treatment-limiting myelotoxicity.[29] Other regimens such as idarubicin, dexamethasone, cytarabine, methotrexate (IDARAM) are also being explored in the HIV setting.
4.4 Summary of Recommendations
|
4.5 References
|
5.0 Cervical Intraepithelial Neoplasia and Cervical Cancer
5.1 Introduction
Cervical cancer is the second most common cancer in women, causing more than 250 000 deaths world-wide in 2005.[1] The majority of these deaths are preventable by systematic cervical screening. The UK National Cervical Screening Programme began in 1988 and, since then, mortality from cervical cancer has fallen dramatically. Peto et al.,[2] in a paper analysing mortality trends before 1988, estimated that without the National Screening Programme one in 65 of all British women born in the UK since 1950 would have now have died of cancer of the cervix (about 6000 women every year). Similar reductions in mortality have been seen in other countries with national screening programmes.[1]
Studies show that the precursor lesion for cervical cancer – cervical intraepithelial neoplasia (CIN) – is more common,[3,4] and more likely to recur,[5,6] in women with HIV infection. Recurrence rates for CIN in HIV-infected women have been estimated to be as high as 56%[7] and up to 87% in severely immunocompromised (CD4 lymphocyte count <200 cells/µL) women.[6] There is also evidence that cervical cancer itself is more common in these women.[8-13] There are mixed data on the effect of highly active antiretroviral therapy (HAART) on CIN, with some studies showing a benefit[14-16] while others do not.[17-19]
Smoking is associated with an increased risk of both CIN and invasive cervical cancer, and the Department of Health currently advocates the promotion of smoking cessation programmes in primary care. These services should be developed in HIV units also, and guidance on the optimal provision of smoking cessation services with particular reference to manual groups, pregnant smokers and hard-to-reach communities is currently being developed by the National Institute for Health and Clinical Excellence (NICE).
In 1993, the Centers for Disease Control and Prevention (CDC) designated invasive cervical cancer in HIV-infected women as an AIDS-defining condition.[20] However, although there are clear data showing an increased prevalence of CIN in women infected with HIV, the data for an increased risk of cervical cancer in these women are mixed. Early studies showed no evidence of a significantly increased risk;[21,22] however, some more recent studies have demonstrated an association between HIV infection and the development of cervical cancer. In the Swiss HIV Cohort Study, Clifford et al.[9] reported a standardized incidence ratio (SIR) of 8.0 [95% confidence interval (CI) 2.9-17.4] for cervical cancer in HIV-infected women over the period from 1993 to 2003, while Frisch et al.[8] in the USA showed a relative risk of 5.4 in women with HIV compared with women without. Several studies from Europe as well as a study from South Africa have shown similar results.[10-13] The increased risk of cancer of the cervix in HIV-infected women appears to be much lower than the increased risk of other HIV-associated malignancies such as Kaposi’s sarcoma (KS) (SIR = 192; 95% CI 170-217) and non-Hodgkin’s lymphoma (NHL) (SIR = 76.4; 95% CI 66.5-87.4).[9] It is interesting to note that there appeared to be no effect of HAART on the prevalence of genital human papilloma virus (HPV) infection in women whose CIN regressed with HAART.[14]
Although some studies have demonstrated a reduction in CIN in women treated with HAART,[15,16] as a consequence of immune reconstitution, none has yet shown a reduction in rates of cervical cancer in women with HIV. The reasons for this are unclear; however, it may be a consequence, in part, of the long transition time between the onset of CIN and the development of cancer. The lack of an effect of HAART on cervical cancer is in contrast to data clearly showing a reduction in KS and NHL in HIV-infected individuals on HAART.[9,15,22] The fact that there appears to be no effect of immune reconstitution in reducing rates of cervical cancer has led Bower et al.[23] to suggest that invasive cervical cancer should no longer be considered an AIDS-defining cancer in the era of HAART.
The recently published UK guidelines for the management of sexual and reproductive health (SRH) of people living with HIV infection produced jointly by the British HIV Association (BHIVA), British Association for Sexual Health and HIV (BASHH) and Faculty of Family Planning and Reproductive Healthcare (FFPRHC) include advice on cervical screening in HIV infection (available online at www.bhiva.org). The key points and recommendations are included below.
In Autumn 2007 the Department of Health (National) recommended that routine HPV vaccination of girls aged 12-13 years should be included in the NHS immunization programme based on advice from the Joint Committee for Vaccination and Immunisation. At present only one quadrivalent vaccine is licensed, Gardasil, which provides protection against HPV genotypes 6, 11, 16 and 18. In the general population genotypes 16 and 18 are responsible for approximately 75% of cervical cancers, 60% of CIN 2/3 and 25% of CIN 1, whilst genotypes 6 and 11 are responsible for 90% of genital warts. Clinical trials have demonstrated that Gardasil can prevent 98% of CIN 2/3, adenocarcinoma in situ or worse.[24,25] No data exist on the efficacy of this vaccine in people with HIV infection; however, it is hoped that HPV vaccination in the future will reduce the burden of HPV-associated disease in this population.
5.2 Key Recommendations of BHIVA, BASHH and FFPRHC 2007 Guidelines on Cervical Screening in HIV
|
5.3 Management of Cervical Intraepithelial Neoplasia
In immunocompetent women, guidelines recommend monitoring of low-grade cervical lesions and surgical treatment of high-grade lesions.[26-28] In HIV-positive women, although the failure rate of surgical excision is higher than in uninfected women and recurrence more likely, the British Society for Colposcopy & Cervical Pathology (BSCCP) recommends that only lesions that are CIN 2 or higher be treated and that women have regular cytological review to detect progression of lower grade lesions which are more likely to be caused by persistent HPV infection and may clear.[26]
Treatment modalities on offer to women with CIN and HIV infection include surgical excision, ablation with electrocautery, cryotherapy or laser, adjunctive medical treatment and hysterectomy. Cautious expectant management may also be indicated. A Cochrane review of 28 randomized controlled trials comparing knife cone biopsy, laser conization, large loop excision of the transformation zone (LLETZ), laser ablation, cryocautery, cold coagulation and radical diathermy showed that none of these techniques was superior to any other for treating or eradicating CIN.[27]
Fruchter et al., in a study of 127 HIV-infected and 193 uninfected women, found that the main risk factors associated with recurrence following treatment were the severity of immunosuppression (assessed by CD4 lymphocyte count) and the presence of residual disease. However, CIN was also found to recur even when no residual disease was evident following treatment. They found that, in HIV-negative women, recurrence rates were low and that, when residual disease was present following treatment, a second excisional procedure permanently removed the disease. These findings were not seen in HIV-positive women in whom CIN was found to persist, recur and progress despite multiple treatments. Recurrence rates in this[6] and another study by Heard et al.[5] were not related to treatment modality. Several studies show that women with HIV are more likely to have incompletely excised margins than immunocompetent women.[6,7] The reasons for this are unclear. At least two studies suggest that cryotherapy may not be as effective at treating CIN as loop excision laser or surgical ablation.[29,30] Expectant management may be considered for early lesions (CIN 1 or lower) as is the case in the general population.[26,31] However, it should be borne in mind that some women may be unsuitable for this approach, particularly those of poor socio-economic status who might have neither the means nor the motivation to seek regular medical follow-up.[32]
Topical 5-fluorouracil (5-FU) has been shown to be an effective adjunct to surgical and ablative therapy for CIN. Maiman et al., in a study of 101 HIV-infected women with CIN 2 and 3 who had undergone excisional or ablative therapy randomly assigned to 6 months of bi-weekly prophylaxis with 5-FU cream (2 g applied vaginally) or observation, found that only 14 out of 50 (28%) women in the 5-FU group developed recurrence after 18 months of follow-up, compared with 24 (47%) of 51 in the observation group.[33] The use of immune modulators is currently undergoing investigation.[34]
5.3.1 The Effect of HAART on Progression of Cervical Intraepithelial Neoplasia. HAART is associated with immune restoration and a reduction in incidence and mortality from opportunistic infections. The effects of HAART on cervical disease are very poorly understood. The use of HAART does not appear to have led to a reduced prevalence of genital HPV infection in women with HIV,[14] although some studies show improved rates of regression of CIN in women on HAART. In the Women’s Interagency HIV Study (WIHS), no CIN lesions regressed in women pre-HAART, but 45% of women on HAART had lesions that regressed to normal cytology with a median time to regression of 2.7 years compared with 59% in HIV-negative women.[15] Heard et al.[16] demonstrated, in a prospective study of 168 HIV-infected women, that CIN lesions regressed in 39.9% of women with a relative hazard of regression of 1.93 (95% CI 1.14-3.29) for women who were receiving HAART compared with women who were not. Studies showing regression of CIN with HAART generally demonstrate a correlation with improving CD4 cell count.
It is important to note, however, that some studies do not show a reduction in CIN with HAART. Orlando et al.[17] failed to show that HAART altered CIN rates in 15 women on HAART who had HPV or low-grade squamous intraepithelial lesions on cervical cytology. In another cohort of 71 women, after 10 months on HAART there was in fact an increase in CIN from 55 to 62%.[18] Lillo et al.[19] also demonstrated no difference in persistence of high-risk HPV or progression of CIN in their study of 163 women.
5.4 Recommendations for Management of Abnormal Smears in HIV-positive Women
|
5.5 Diagnosis, Staging and Prognosis of Invasive Cervical Cancer
The diagnosis of invasive cervical cancer may be suggested by the finding of an abnormal cervix on vaginal or speculum examination and should be confirmed on histology of tissue specimens. The positive predictive value of cervical cytology in predicting biopsy-proven CIN 3 or worse is estimated at 56% for CIN 2-3 and 4% for invasive cancer.[35] The staging of cancer of the cervix is clinical rather than based on imaging (see Table 8 ). This is because of the international significance of this cancer and the lack of widespread availability of computed tomography (CT) and magnetic resonance (MR) scanning. However, MR scanning has been evaluated as an adjunct to clinical staging and found to be useful.[36]
Baseline evaluation should include vaginal and rectal examination, colposcopy, cystoscopy, endocervical curettage, hysteroscopy, intravenous urogram, and chest and skeletal X-rays. Information gained from lymphangiography, ultrasonography, CT and MR scanning and laparoscopy is useful in planning treatment but is not generally used in staging. Routine haematological and biochemical parameters should also be assessed.
The prognosis for patients with cervical cancer is markedly affected by the extent of disease at the time of diagnosis. Dissemination of carcinoma of the cervix is by invasion of the connective tissue stroma and thence by contiguous spread to the adjacent parametrial tissue and beyond with involvement of the regional lymph nodes. Involvement of the ureters may result in hydronephrosis and renal failure. Patients may present with symptoms and signs pointing to involvement of local or distant organs: invasion of the sciatic nerve roots may cause back pain, and pelvic vein and lymph node involvement may result in oedema of the lower limbs. Haematogenous spread may occur without nodal involvement. Distant metastases occur late, with involvement of the para-aortic lymph nodes, lungs, liver and bone. Important prognostic factors include stage, volume and grade of tumour, histological type, lymphatic spread and vascular invasion.
Data from a surgicopathological staging study of patients with clinical stage IB disease showed that the most important predictive factors for lymph node spread metastases and a reduced disease-free survival time were involvement of the capillary-lymphatic space, increasing tumour size and increasing depth of stromal invasion.[37,38] Another study involving 1028 patients treated with radical surgery showed a greater correlation of survival with size of tumour than with clinical or histological stage.[39] A further study found that other factors associated with progression-free interval and survival were age of patient, lack of involvement of para-aortic and pelvic lymph nodes, unilateral disease and performance status. This study, however, did find a correlation with clinical stage.[40] It is unclear whether cervical adenocarcinoma of the cervix carries a significantly worse prognosis than squamous cell carcinoma of the cervix.
5.6 Management of Invasive Cervical Cancer
Despite widespread cervical screening, data from the Surveillance, Epidemiology and End Results (SEER) programme of the US National Cancer Institute show that up to 50% of women present with stage II-IV disease.[41] Concurrent treatment with cisplatin-based chemotherapy and radiotherapy has been shown in randomized phase III trials to offer a survival advantage over radiation therapy in combination with surgery or alone. Mortality from cervical cancer was reduced by 30-50% with the use of combination chemotherapy and radiotherapy.[42-47]
5.6.1 Stage IA.
|
5.6.2 Stage IB/IIA. The options for treatment of stage IB disease are as follows.
|
5.6.3 Stage IIB/III/IVA. Radiation therapy plus chemotherapy with cisplatin or cisplatin/5-FU for patients with bulky tumours is standard treatment[42-47] using external-beam pelvic radiation therapy combined with two or more intracavitary brachytherapy applications. HDR brachytherapy is now being increasingly used, typically with 192-Ir. This gives the advantage of eliminating radiation exposure of medical personnel, a shorter treatment time, patient convenience and out-patient management. In three randomized trials, HDR brachytherapy was comparable to LDR brachytherapy in terms of local-regional control and complication rates.[52-57]
5.6.4 IVB/recurrent Cancer. There is no standard regimen for stage IVB or recurrent disease that provides substantial palliation. Individuals with these conditions should be enrolled into appropriate clinical trials where possible. Radiotherapy may be considered for palliation of central or metastatic disease.
5.6.5 Invasive Cancer in Pregnancy. The general recommendations are that invasive cancer should be treated according to the stage of disease and gestational age of the foetus at diagnosis. When disease is diagnosed before foetal maturity, immediate treatment is usually indicated, and for disease detected in the third trimester, therapy may be delayed until after delivery.[58,59] It should be noted that some reports suggest that delaying treatment to improve foetal outcome may also be an option in early disease (IA and early IB).[60-62]
5.7 Summary of Recommendations for Management of Women with Cervical Cancer
Stage IA
|
Stage IB/IIA
|
Stage IIB/III/IVA
|
Stage IVB/recurrent disease
|
5.8 References
|
6.0 Anal Cancer
6.1 Introduction
The recently published UK guidelines for the management of the sexual and reproductive health (SRH) of people living with HIV infection, produced jointly by the British HIV Association (BHIVA), BASHH and FFPRHC, include advice on anal cancer in HIV infection (available online: www.bhiva.org). The key points and recommendations are included below.
6.2 Key Recommendations of BHIVA, BASHH and FFPRHC 2007 Guidelines on Anal Cancer in HIV
|
6.3 Key Recommendations of NICE 2004 Guidelines on Anal Cancer
|
6.3.1 Primary Treatment. Concurrent chemoradiotherapy, using mitomycin C, 5-fluorouracil and radiation, is appropriate for most patients. Other forms of treatment, such as surgical excision, may be considered by anal cancer multidisciplinary teams (MDTs), but surgery is usually reserved for salvage. There are still some areas of uncertainty about optimum treatment, and eligible patients should be encouraged to participate in trials such as the Cancer Research UK (CRUK) ACT 2 trial.
6.3.2 Management of Relapse. All patients with suspected or confirmed relapse should be discussed by the anal cancer MDT. Those with confirmed locoregional recurrence should undergo cross-sectional imaging and all treatment options, including surgery, should be considered by the MDT. Palliative radiotherapy, chemotherapy and palliative care should be discussed with patients who have metastatic disease or who are not sufficiently fit to undergo potentially curative treatment.
6.4 Diagnosis, Staging and Prognosis of HIV-associated Anal Cancer
The incidence of anal carcinoma amongst people with HIV[2] and men who have sex with men (MSM) is markedly increased.[3-5] Moreover, anal cancer is twice as common in HIV-positive MSM as it is in HIV-negative MSM.[6] US AIDS cancer registry matching calculated that the relative risk of invasive anal cancer is 37 in HIV-positive men and 6.8 in HIV-positive women.[7] There is no apparent correlation between the relative risk of developing invasive anal cancer and the CD4 cell count,[7,8] although trends have been observed.[9] In addition, highly active antiretroviral therapy (HAART) can lead to regression of Kaposi’s sarcoma and cervical intraepithelial neoplasia[10] but does not appear to lead to resolution of anal intraepithelial neoplasia (AIN). Furthermore, the duration of immune dysfunction (as measured by the interval from HIV infection to anal cancer diagnosis) is longer in patients who have developed anal cancer in the era of HAART.[11-13] For example, in a cohort study, the overall incidence of invasive anal cancer was 60 per 105 patient-years [95% confidence interval (CI) 40-89]. This compares to an incidence of 0.52 (95% CI 0.27-0.78) per 105 patient-years in the age- and gender-matched general population of southeast England. Moreover, the incidence of invasive anal cancer in the HIV-positive cohort has not declined since the introduction of HAART. The incidence was 35 (95% CI 15-72) per 105 patient-years of follow-up in the pre-HAART era and is 92 (95% CI 52-149) per 105 patient-years of follow-up in the post-HAART era.[14,15]
Squamous cell (epidermoid) carcinomas make up the majority of all primary cancers of the anus. The anal canal extends from the rectum to the perianal skin and is lined by a mucous membrane that covers the internal sphincter. The following is a staging system for anal canal cancer that has been described by the American Joint Committee on Cancer (AJCC) and the International Union Against Cancer.[16] Tumours of the anal margin (below the anal verge and involving the perianal hair-bearing skin) are classified with skin tumours. Staging involves a CT scan of the chest, abdomen and pelvis, and ideally an MRI of the pelvis. Anal cancer is staged according to the Tumour, Nodes and Metastases (TNM) definitions (see Table 9 ).
A significant problem has been the relatively advanced stage of disease at presentation, with 38% having T3 or T4 disease, 31% having nodal disease and 6% presenting with distant metastases.[14,15] This late presentation can be explained in part by the attribution, in the clinical setting, of anal symptoms to the presence of warts or haemorrhoids. Advanced presentation of warts may be a reflection of more aggressive disease in HIV-positive patients rather than a failure to diagnose early. This has not helped MSM to be aware of the potential seriousness of a lump in the anus, requiring biopsy. Most centres report that in the general immunocompetent population 85% of tumours can be controlled locally with chemoradiotherapy and 5-year survival rates are in the range of 65-85%. The overall actuarial survival at 2 years for HIV-positive patients with invasive anal cancer is 47% (95% CI 24-70%).[14,15] A comparison of 10 HIV-seropositive patients with anal cancer receiving HAART with 10 age-matched seronegative patients who were treated with 3D conformal radiotherapy of 59.4 Gy and standard 5-fluorouracil and mitomycin C showed that overall survival at 5 years was 70% in HIV-seropositive patients receiving HAART and 69% in the matched controls. Colostomy-free survival was 70% (HIV-positive) and 100% (matched HIV-negative). No HIV-seropositive patient received an interstitial brachy-therapy boost compared with 42% of HIV-seronegative patients, and adherence to chemotherapy seemed to be difficult in HIV-seropositive patients. Acute haematological toxicity was high in HIV-seropositive patients, reaching 50%, compared with 12% in HIV-seronegative patients, but the rate of long-term side effects was low in HIV-seropositive patients.[17]
6.5 Management of Anal Cancer
For anal cancer, local control and sphincter preservation, as for immunocompetent individuals, remain the major challenges.[18] Abdominoperineal resection leading to permanent colostomy was previously thought to be required for all but small anal cancers below the dentate line, with approximately 70% of patients surviving 5 or more years in a single institution,[19] but such surgery is no longer the treatment of choice.[20] Radiation therapy alone may lead to a 5-year survival rate in excess of 70%, although high doses (≥60 Gy) may yield necrosis or fibrosis, toxicities that are probably greater in HIV-positive patients. Chemotherapy concurrent with lower dose radiation therapy has a 5-year survival rate in excess of 70%, with low levels of acute and chronic morbidity, and few patients require surgery for dermal or sphincter toxic effects. The optimal dose of radiation with concurrent chemotherapy to optimize local control and minimize sphincter toxic effects is under evaluation but appears to be in the 45-60 Gy range. Analysis of an intergroup trial that compared radiation therapy plus fluorouracil/mitomycin with radiation therapy plus fluorouracil alone in patients with anal cancer has shown improved results (lower colostomy rates and higher colostomy-free and disease-free survival) with the addition of mitomycin.[21] Standard salvage therapy for those patients with either gross or microscopic residual disease following chemoradiotherapy has been abdominoperineal resection. Alternatively, patients may be treated with additional salvage chemoradiotherapy in the form of fluorouracil, cisplatin and a radiation boost to potentially avoid permanent colostomy.[21]
In summary, for anal cancer, in phase II studies in HIV-positive individuals, the best outcomes appear to have derived from the use of combined modality therapy of radiotherapy and concurrent chemotherapy.[17,22-26] This generally has involved 5-fluorouracil and mitomycin C, and concomitant radical radiotherapy to the pelvis (38-51 Gy in 20-30 fractions), with most patients receiving a perineal boost (10-18 Gy). The commonest grade 3 toxicities are haematological, gastrointestinal and skin (all >20%) although, in general, radical chemoradiation may be given safely at conventional doses in HIV-positive patients, with a complete response rate of >80% in those with stage I-III disease.[23,24] There is no evidence that HAART can cause regression of anal cancer, but we recommend its use to prevent other infections, maintain CD4 cell count and suppress viraemia.[14,15]
6.6 Screening for Anal Intraepithelial Neoplasia
Despite an extensive understanding of the biology of human papilloma virus (HPV), the aetiological agent of anal cancer, the relationship between AIN and invasive cancer remains poorly understood. Treatment options for AIN are limited by morbidity and high recurrence rates and there are no randomized trials studying the efficacy of therapeutic agents or surgery for high-grade AIN (HG-AIN), although immunotherapies show early promise. Theoretically, early detection may lead to better treatment outcomes and studies of the potential negative consequences of screening programmes on MSM populations are also required. Using widely accepted criteria for the introduction of screening programmes, there is little evidence for its routine use as the early detection of lesions still poses substantial difficulties,[27] and single biopsies may miss areas of AIN, with histology and cytology yielding some discordant results.[12] In a comparison between results of anal cytology using the sampling method of Palefsky and histological findings of biopsies taken from abnormal areas seen on direct high-resolution anoscopic examination of the anal canal, the sensitivity of the cytology was 82%, and the specificity 45% when compared with histology. Of the patients with no detectable AIN, 77% had a high-risk HPV type in the anal canal, rising to 94% in patients with HG-AIN, thought to be the precursor of anal cancer. There were no significant differences in the prevalence of HPV-16 or all high-risk HPV genotypes between different cytological or histological grades of abnormalities.[27] The utility of an AIN/anal cancer screening programme is currently being investigated. All patients diagnosed with AIN recently are being followed regularly (by the rectal clinic) but not with cytology and, based on current evidence, we cannot recommend cytological screening. Recommendations include increased awareness and education for HIV-treating clinicians, lower threshold for referral for biopsy for patients with anal symptoms and regular review (including clinical or anoscopy or cytology or histology) for patients with known AIN. This is also based on the fact that treatment responses are often poor, because of late diagnoses.[11,18-24]
AIN may be treated with topical imiquimod, an immunomodulator, and/or surgery, as well as regular follow-up, although there is no standard treatment.[28] Patients should receive the optimal HAART regimen. The level I evidence associated with use of HPV vaccines to prevent cervical cancer has yet to be applied to HIV-associated anal cancer. Services should probably employ specialist staff full-time to deal with this workload. More evidence will need to be provided in this climate.
6.7 Summary of Guidance
|
6.8 References
|
7.0 Hodgkin’s Lymphoma
7.1 Introduction
Hodgkin’s lymphoma (HL) is one of the commonest tumours amongst the non-AIDS-defining malignancies (non-ADM)[1,2] with a 10- to 20-fold increased incidence in HIV-infected patients in comparison with the HIV-negative population.[1,3-6] Conflicting results have been reported regarding the incidence of HL after the advent of highly active antiretroviral therapy (HAART): some authors have reported a slight increase in HL incidence,[6] whereas others have not detected any difference in the incidence of HL in the pre-HAART and post-HAART eras.[7,8]
HL in HIV-infected patients tends to present more frequently in advanced stage at diagnosis, with extranodal involvement, especially bone marrow infiltration, and with a higher proportion of patients with B symptoms and poor performance status than in the general population.[9-12] From a histological point of view, HL in HIV-infected patients is characterized by a predominance of the mixed cellularity (MC) and lymphocyte depleted (LD) subtypes, as opposed to nodular sclerosis (NS),[5,9-11,13,14] and by a higher percentage of Epstein-Barr virus (EBV) positivity.[9,11]
The prognosis of HIV-HL in the pre-HAART era was considerably worse than in HIV-negative patients, with complete remission (CR) rates ranging from 44 to 65%,[9,13,15,16] and median overall survival (OS) of about 18 months.[9,15,16] However, the outcome of HIV-infected patients with HL has dramatically improved since the introduction of HAART; the CR rate, OS and disease-free survival (DFS) approach those seen in the general population,[12,16] although the results of treatment for HL in HIV-infected patients are still worse than in HIV-negative patients.
7.2 Diagnosis, Staging and Prognostic Factors
The diagnosis of HL, as that of any other lymphoid malignancy, should be based on a tissue sample biopsy, rather than on a cytological sample. Samples should be stained for CD20, CD3, CD15, CD30, B cell lymphoma-2 (BCL-2) and latent membrane protein-1 (LMP-1) proteins.
Following the confirmation of diagnosis, patients should undergo a series of investigations [including blood tests, body computed tomography (CT) scan and unilateral bone marrow biopsy] to assess the extension of the disease (see Table 10 ). Whereas a bone marrow biopsy is not necessary in all HIV-negative patients with HL, the higher proportion of bone marrow involvement in the HIV-infected population[9,15] makes it mandatory. The above-mentioned investigations allow staging of the disease according to the Ann Arbor classification/Cotswolds modification[17] (see Table 11 ).
A prognostic score, which predicts both freedom from progression (FFP) and OS, has been defined for HIV-negative patients with advanced HL at diagnosis[18] (see Table 12 ). The applicability of the International Prognostic Score (IPS) in HIV-infected patients was reported in a series of patients treated with Stanford V chemotherapy, in which the IPS was the only variable predictive for OS in the multivariate analysis. The IPS also predicted for FFP and CR rate.[19] Other prognostic markers that have been reported to have an impact on the outcome of HIV-HL patients include some predictive factors related to characteristics of the lymphoma, such as age, stage and responsiveness to therapy,[12,20] and others associated with the HIV infection and/or its treatment.[12,16,20-22] Histological subtypes have been associated with prognosis in the HIV-infected population in some studies[21] but not in others.[20]
The prognostic impact of treatment with HAART on the outcome of HIV-HL has been addressed in a few studies comparing either the prognoses of patients in the pre- and post-HAART eras or the outcomes of those actually treated with HAART and those not receiving HAART. Ribera et al.[21] reported a higher CR rate, longer OS and longer DFS amongst patients who were receiving HAART when they were diagnosed with HL or started HAART at the same time of diagnosis, in comparison with patients who did not receive HAART before or during HL treatment. In this series, treatment with HAART emerged as an independent prognostic factor for response, survival and DFS in the multivariate analysis. A single-centre French study including 108 patients reported a higher, although not statistically significant, CR rate for patients in the post-HAART era and for those actually receiving HAART. The OS and DFS were significantly longer in the post-HAART era and in patients treated with HAART.[16] In a multicentre German study, response to HAART and age were associated with a prolonged OS in the multivariate analysis.[12] Similarly, another multicentre German study identified treatment with HAART as an independent prognostic factor for OS, along with stage, CD4 cell count and response to chemotherapy.[20] Based on these results, and taking into account the impossibility of testing the efficacy of HAART in a randomized trial, it is recommended to start HAART in patients diagnosed with HL.
7.3 Re-assessment and Follow-up
Specific response criteria for HL in HIV-infected patients have not been described, but the response criteria defined for the general population are generally used. In some recent series[23] the recommendations of the International Working Group for assessment of response published in 1999[24] were followed. The mentioned guidelines were developed for patients with non-Hodgkin’s lymphoma (NHL) and they have been recently reviewed and updated to include HL, amongst other modifications.[25] One of the important changes in the guidelines is that they include positron emission tomography (PET) scan for the assessment of residual masses in HL. The literature on PET scan in HIV-lymphoma is mostly limited to its role in distinguishing primary central nervous system lymphoma (PCNSL) from infection in patients with central nervous system (CNS) lesions. In the absence of specific data on the applicability of PET scan to assessing the response in patients with HIV-HL, the same response criteria as in HIV-negative patients should be followed. Thus, assessment after treatment should include Neck Chest Abdomen Pelvis (NCAP) CT scans and a bone marrow biopsy (if the CT scan shows CR and the bone marrow was involved at diagnosis). Patients with a residual mass should have a PET scan. These investigations should be performed at least 4-6 weeks after the last cycle of chemotherapy.
Regarding follow-up, it is recommended for patients in CR after treatment to have an oncology appointment every 3 months for the first and second years, every 6 months for the following 3 years and then annually. Investigations at follow-up should include medical history, physical examination and blood tests. No further surveillance investigations are recommended for patients in CR. The small group of patients who have received radiotherapy (RT) should have thyroid function tests (TFT) checked regularly and an annual chest X Ray (CXR) if they have had mediastinal RT, and female patients treated with Mantle RT should have surveillance (mammographies/magnetic resonance imaging).
7.4 Management
7.4.1 First-line Treatment. No randomized studies have addressed the question of the best chemotherapy regimen for patients with HL and HIV infection, and the data are derived mainly from non-randomized controlled trials or case series (see Table 13 ).
In the pre-HAART era, Errante et al. included 17 patients with advanced-stage HL (stages I-II with adverse prognostic factors or stages II-IV) in a trial analysing the impact of adding zidovudine (ZDV) to the combination chemotherapy EBV (epirubicin, bleomycin and vinblastine). The patients were stratified according to performance status and presence of previous opportunistic infections (OIs) to receive either the full dose or a reduced dose of chemotherapy. The CR rate in the whole group was 53%, with six of nine patients remaining in CR at a range of 12-37 months. One patient developed tuberculosis after the third cycle but no other OIs were observed[26] ( Table 14 ).
The European Intergroup Study added granulocyte colony-stimulating factor (G-CSF) to EBVP (epirubicin, bleomycin, vinblastine and prednisone) chemotherapy in an attempt to improve these results. Patients received either ZDV or didanosine (ddI) as antiretroviral therapy (ART). The CR rate was higher than in the EBV study (74%), but the median OS was similar (16 months). No significant differences were found when results with EBVP were compared with those of the EBV study, and the authors concluded that the addition of G-CSF did not result in an apparent advantage.[22]
Another series investigated the addition of G-CSF to standard chemotherapy (ABVD: doxorubicin, bleomycin, vinblastine and dacarbazine). In this study the patients did not receive any ART. In spite of the use of G-CSF, a considerable proportion of patients developed grade 3-4 neutropenia and one-third of the patients developed OIs. Of note, in this study the CD4 cell count at diagnosis of HL was lower than in other series (median CD4 cell count 113 cells/µL). With regard to the response to chemotherapy, the results were similar to those previously reported (CR 43%; median OS 18 months).[27]
In contrast, a small Italian series reported much better results with the same protocol. Eight patients were treated with ABVD and G-CSF from 1994 to 1999. The CR rate in this study was 100%, with a median OS of 43 months. One patient died in CR 46 months after the diagnosis of HL and the rest remained in CR (range 31-96 months). The toxicity reported was slightly less severe than in other series. The patients in this series had a higher CD4 cell count than in the Levine et al. series, there was a lower percentage of patients with AIDS at the time of HL diagnosis (12.5%), and two of them received HAART while on chemotherapy.[27]
ABVD remains the standard of care for HIV-negative patients with HL.[29] Although there is a suggestion that more intensive chemotherapies result in higher response rates,[30] there has not been demonstrated a survival advantage for these intensive chemotherapies over ABVD in a randomized study. An ongoing trial is addressing the question. In the setting of HIV infection, some groups have investigated the use of such more intensive chemotherapies in the HAART era.
Spina et al. treated 59 patients with a Stanford V chemotherapy regimen with G-CSF support, concomitantly with HAART. Approximately two-thirds of the patients managed to complete the 12-week treatment but 31% required a dose reduction. One treatment-related death as a result of septic shock and four episodes of OIs were reported. In addition to myelotoxicity, nonhaematological toxicity was also more severe than with less intensive chemotherapies, with 54% of patients developing neurotoxicity. In terms of response to chemotherapy, CR was achieved in 81% of the patients with a 3-year OS of 51%. Fifty-six per cent of the patients were alive and free of disease after a median follow-up time of 17 months, with a 3-year DFS of 68%.[31]
A multicentre pilot study reported the use of the intensive chemotherapy Bleomycin, Eroposide, Adriamycin, Cyclophosphamide, Vincristine, Procarbazine, Prednisolone (BEACOPP) in patients with HL and HIV infection. Amongst 12 patients included in the study, four discontinued the treatment before completing the six cycles, because of OIs in two and prolonged neutropenia in the remainder. G-CSF, which was optional, was given in 54% of the cycles. In spite of this, grade 3-4 neutropenia was seen in 75% of patients, with six episodes of grade 3-4 infection (including one patient requiring intensive care and one patient with pneumonia). CR was achieved in all patients (in one case after RT) and nine remained in CR at a median of 49 months (range 13-108 months).[23]
In the absence of randomized trials showing an advantage of the more intensive regimens BEACOPP and Stanford V over ABVD (either in HIV-infected patients or in the HIV-negative population), and given their toxicity, these intensive chemotherapies should probably not be used outside the setting of controlled trials.
Regarding the use of RT in the treatment of HIV-HL, not much information is available, because of the small proportion of patients diagnosed with localized disease or with mediastinal disease, on the one hand, and because of concerns regarding its toxicity when given after chemotherapy in this population, on the other hand. In the Rubio et al.[15] study, three patients with localized disease were treated with local RT but none of the patients with advanced stage at diagnosis received RT as part of the treatment. Similarly, in the Errante et al.[13] series, a significantly lower proportion of HIV-positive patients received combined treatment with chemotherapy and RT in comparison with the general population. Amongst 10 patients eligible for RT in the Stanford V study [partial response (PR) after chemotherapy or CR in patients with bulky disease at diagnosis], only six received the planned treatment and no data on them were reported.[31] Two patients in the BEACOPP study received RT because of bulky and residual disease, respectively. The patient in PR achieved CR after RT but, again, no details on the toxicity were reported.[23] Thus, no conclusions can be drawn regarding the use of RT in HIV-infected patients.
7.4.2 Treatment at Relapse. The standard treatment of relapsed/refractory HL in the HIV-negative setting is salvage chemotherapy followed by high-dose chemotherapy and stem cell transplantation. No series have been published on the salvage treatment in HIV-infected patients with relapsed/refractory HL and, thus, the scarce available data are extracted from series on first-line treatment or studies of high-dose treatment (HDT) with stem cell rescue (SCR). The salvage protocols used are varied and include ABVD, Mechlorethamine, Vincristine, Procarbazine, Prednisolone (MOPP), Cyclophosphamide, Mechlorethamine, Vincristine, Procarbazine, Prednisolone (CMOPP)-ABV, MOPP/ABV, Cyclophosphamide, Vincristine, Procarbazine, Prednisolone (COPP)-ABV, BEACOPP, vinorelbine, Eroposide, Methylprednisolone, Cytarabine, Cisplatin (ESHAP), Mitoguazone, Ifosfamide, Vinorelbrine (MINE), ifosfamide-VP16, ifosfamide-VP16-mitoxantrone and RT.[22,32-34] Seven patients out of 10 treated with EBVP who relapsed after CR and two out of six in PR received rescue chemotherapy. Only one of the relapsed patients achieved a PR that lasted 18 months after salvage MOPP/ABV and one PR patient achieved an ongoing CR 9 months after receiving MOPP. The rest of the patients died with disease progression.[22]
A few series have analysed the feasibility of HDT with SCR in patients with HIV and lymphoma ( Table 14 ). Re et al. reported the outcomes of 10 patients with lymphoma, including four with HL, receiving HDT with SCR. A considerable proportion of the patients in this series had advanced/refractory disease. Four episodes of OIs were seen after HDT (two cases of oesophageal candidiasis and two of varicella zoster), but no patients died of OIs or treatment-related complications.[35]
In the Gabarre et al. series, six patients with HL were included, three treated at first relapse, and one each treated at second, third and fourth relapses. The toxicity was similar to that reported in HIV-negative patients, with febrile neutropenia as the most frequent complication and no transplant-related mortality (TRM). Two patients developed cytomegalovirus (CMV) reactivation which was treated with ganciclovir (one of them developed pancytopenia after the treatment, which persisted for 1 year, and died of invasive aspergillosis 16 months after HDT). Three patients with HL remained in CR after 19, 32 and 49 months, respectively.[32]
In the Serrano et al. series, three patients received HDT for relapsed HL: two were in second CR and one in PR at the time of treatment. The latter developed disease progression and died at day 19 of HDT whereas one patient remained in CR at 17 months and the other relapsed 32 months after HDT. No deaths related to HDT were reported but toxicity was slightly worse than that seen in HIV-negative patients: 63% of patients needed intravenous nutrition because of severe mucositis. The following OIs were reported: CMV infection, disseminated herpes zoster and varicella zoster.[33]
Krishnan et al. published the results of HDT in 20 HIV-infected patients with lymphoma including two with HL, who remained in CR 56 and 61 months after HDT. One of the patients in this series, who was 68 years old, developed cardiomyopathy and renal failure and died at day 22 of HDT. Other toxicities reported were engraftment syndrome, interstitial pneumonitis, pericarditis, supraventricular arrhythmia, subdural haematoma and haemorrhagic cystitis. With regard to OIs post-HDT, eight episodes were reported: two of pneumocystis pneumonia, one of pulmonary aspergillosis, two of disseminated herpes zoster, one of CMV retinitis and two of CMV reactivations.[34]
Although the series are small and very heterogeneous in terms of the patients included (NHL and HL; patients in first CR and others with refractory disease), and the follow-up is still limited, it can be concluded from them that, in patients who have remaining antiretroviral options to control their HIV infection long-term, HDT, although still experimental, is a valuable option. All the series showed that the CD4 cell count remained stable (or slightly decreased) and the viral load remained unchanged in patients receiving HAART during HDT. The majority of the patients managed to continue receiving HAART while having HDT with occasional short interruptions because of gastrointestinal intolerance or mucositis.
7.5 Recommendations
|
7.6 References
|
8.0 Multicentric Castleman’s Disease
8.1 Introduction
Multicentric Castleman’s disease (MCD), a relatively rare lymphoproliferative disorder that presents with fevers, anaemia and multifocal lymphadenopathy, is today most commonly diagnosed in individuals infected with HIV type 1. The first description of Castleman’s disease appeared as a case record of the Massachusetts General Hospital in the New England Journal of Medicine in 1954.[1] Benjamin Castleman, the pathologist at Massachusetts General Hospital, subsequently described 13 cases of asymptomatic localized mediastinal masses demonstrating lymph node hyperplasia resembling thymoma in 1956.[2] The localized form usually presents in young adults with isolated masses in the mediastinum (60-75%) or neck (20%) or less commonly intra-abdominal masses (10%). Systemic symptoms are rare with localized Castleman’s disease (LCD). In contrast, MCD presents with polylymphadenopathy and frequently multi-organ involvement, is associated with systemic features, and follows a more aggressive natural history. MCD is predominantly of the plasma cell variant characterized by large plasmablasts in the mantle zone.
In general, MCD occurs in the fourth or fifth decade of life but at younger ages in people who are HIV-positive. Patients often present with generalized malaise, night sweats, rigors, fever, anorexia and weight loss. On examination, they have multiple lymphadenopathy, hepatosplenomegaly, ascites, oedema and effusions both pulmonary and pericardial. Laboratory investigations may reveal thrombocytopenia, anaemia, hypoalbuminemia and hypergammaglobulinemia. Patients may also present with pancytopenia and organ failure (particularly respiratory and renal), as well as with shock requiring admission to intensive care units. MCD is more likely to lead to neuropathic complications than locally confined Castleman’s disease. Patients can develop polyneuropathies and leptomeningeal and central nervous system (CNS) infiltration as well as myasthenia gravis.[3] The polyneuropathy is a chronic, inflammatory demyelinating neuropathy and may be present as part of the rare POEMS syndrome (Crow-Fukase disease). POEMS syndrome consists of polyneuropathy, organomegaly, endocrinopathy monoclonal gammopathy and skin changes. Patients are diagnosed with POEMS syndrome if they have two of these clinical features as well as plasma cell dyscrasia.
MCD is a relapsing and remitting disease and the definition of an ‘attack’ has recently been proposed as a combination of fever and a raised serum C-reactive protein in the absence of other aetiology plus three of the following symptoms: peripheral lymphadenopathy, splenomegaly, oedema, pleural effusion, ascites, cough, nasal obstruction, xerostomia, rash, central neurological symptoms, jaundice or autoimmune haemolytic anaemia.[4]
There is an association between MCD and AIDS-associated Kaposi’s sarcoma (KS), which was also recognized again following initial publication of case reports.[5] In 1994, Chang and Moore isolated a new human gamma herpesvirus from AIDS-KS lesions using differential representational analysis.[6] This virus, known as human herpesvirus 8 (HHV8) or Kaposi’s sarcoma herpesvirus (KSHV), was later found to be present in all cases of HIV-associated MCD[7] and many of the features of this condition can be explained by its presence.
8.2 Diagnosis
The first step towards successfully making the diagnosis of MCD is to consider it in high-risk patients. The diagnosis of MCD is established histologically by lymph node biopsy or if necessary splenectomy. The characteristic features of HIV-associated MCD are interfollicular plasmablasts that express the HHV8 latent nuclear antigen (LANA). These plasmablasts also express high levels of λ light-chain restricted immunoglobulin M (IgM), but are polyclonal and do not contain somatic mutations in their IgV genes, suggesting that they arise from naïve B lymphocytes.[8] Occasionally these plasmablasts join together to form clusters or ‘microlymphomas’ and may progress to monoclonal plasmablastic lymphomas.[9] HHV8 is also present in the malignant cells of these plasmablastic lymphomas.[10,11] HHV8 encodes a viral homologue of interleukin-6 (vIL-6) as a lytic virokine. Only 10-15% of HHV8-positive plasmablasts in MCD express vIL6; however, the human IL-6 receptor is expressed by all HHV8-positive plasmablasts. It is hypothesized that activation of the IL-6 signalling pathway by HHV8 vIL-6 may transform naïve B cells into plasmablasts and lead to the lymphoproliferative diseases associated with this virus, including MCD.
Once the diagnosis has been established, further work-up including laboratory tests and imaging is warranted. Laboratory studies should include testing for HHV8 DNA in plasma or from peripheral blood mononuclear cells by real-time polymerase chain reaction (PCR). Preliminary studies suggest that plasma HHV8 viral load may be a usable tumour marker in HIV-associated MCD, helping in the diagnosis of MCD and in monitoring of responses to treatment and in the diagnosis of relapses.[4,12]
8.3 Staging
Following diagnosis, patients should have a staging neck, chest, abdomen and pelvis computed tomography (CT) scan to assess the extent of disease, and monitor response to treatment. It is unclear whether a bone marrow biopsy to exclude microlymphoma should be recommended. The role of functional imaging such as fluorodeoxyglucose positron emission tomography (FDG-PET) scans is uncertain.
8.4 Prognosis
HIV-associated MCD is relatively uncommon and only recently recognized, so the incidence and prognosis are not well established. The effect of the HAART era on incidence and prognosis is similarly unclear. Not only is MCD itself potentially fatal as a result of organ failure but it is also associated with an increased incidence of non-Hodgkin’s lymphoma (NHL). In a prospective study of 60 HIV-infected individuals with MCD, 14 patients developed HHV8-associated NHL. Three patients had classic HHV8-positive, Epstein-Barr virus (EBV)-positive primary effusion lymphoma (PEL); five were diagnosed with HHV8-positive/EBV-negative visceral large B-cell lymphoma with PEL-like phenotype, and six developed plasmablastic lymphoma/leukaemia.[11] This is a 15-fold increase in lymphoma risk above that seen in the HIV-infected population. The pathogeneses of these lymphomas probably differ, with the plasmablastic type driven by the expansion of plasmablastic microlymphomas seen in MCD lesions. In contrast, the PEL and PEL-like lymphomas may be driven by the cytokine-rich environment with high levels of IL-6 and IL-10, which are known to enhance cell growth of PEL cell lines.[13]
8.5 Management
There are no definitive gold-standard treatments for MCD. No randomized trials have been conducted on account of the infrequency of the diagnosis and often only case reports have appeared in the literature.
8.6 Surgery
Although surgery is the mainstay of treatment for LCD, with complete removal of the mediastinal lesions being curative, it has a limited role in MCD. Splenectomy, in addition to establishing the histological diagnosis, may have a therapeutic benefit as a debulking procedure, as some of the haematological sequelae such as thrombocytopenia and anaemia may in part be caused by splenomegaly. Following splenectomy there is often resolution of the constitutive symptoms but this may be short-lived, approximately 1-3 months, and some form of maintenance therapy is needed to prevent relapse.[14]
8.7 Chemotherapy
For immunocompetent patients the chemotherapy regimens for MCD are based on lymphoma schedules such as CHOP (cyclophosphamide, doxorubicin, vincristine and prednisolone).[14] Although there is little evidence on which to base treatment strategies in HIV-associated MCD, many centres use single-agent chemotherapy with vinblastine or etoposide to induce remission in aggressive forms of MCD. This may be followed by maintenance treatment with these agents.[15]
8.8 HAART
The effect of HAART, chiefly in combination with cytotoxic chemotherapy, has been described in seven patients with MCD and HIV infection.[16] Six patients responded to chemotherapy, and immune reconstitution was described in five patients. However, patients continued to require long-term maintenance chemotherapy to prevent further episodes of MCD. The mean survival was 48 months, which was longer than described in the pre-HAART era when most patients succumbed to opportunistic infections related to their HIV infection.
8.9 Immunotherapy (excluding rituximab)
Specific immunotherapy has also been used as treatment for MCD. Interferon alpha (IFN-α) has been administered either alone or in combination with HAART or chemotherapy for patients with MCD both to induce remission and as maintenance therapy.[14,17,18] In combination with vinblastine and splenectomy, IFN-α contributed to the long-term remission of two of three patients.[14] In a case report a patient was initially treated with antiviral therapy and splenectomy followed by chemotherapy to induce remission and, although this was achieved, chemotherapy failed to achieve sustained remission and IFN-α therapy was started.[18] This has led to remission for over a year. A further case report of treatment of MCD with HAART and low-dose IFN-α alone has shown a sustained remission of 24 months.[19]
As the pathogenesis of MCD is related to HHV8 virus and its viral oncogenes, particularly vIL-6, monoclonal anti-IL-6 therapy has also been used in the treatment of MCD. Seven HIV-negative patients were treated with atlizumab, a humanized monoclonal anti-IL-6 receptor antibody in patients with either multicentric plasma cell or mixed variant Castleman’s disease. They had resolution of their immediate symptoms and, by 3 months, they had reduction in lymphadenopathy and hypergammaglobulinemia with improvement of their renal function, which had deteriorated as a result of secondary amyloidosis. This remission was not sustained and recurrence was observed.[20] These studies have been expanded to a multicentre clinical trial in Japan[21] but there are no reports of the use of atlizumab in persons with HIV. Recent case reports of treatment with thalidomide also showed resolution of systemic manifestations of MCD, and the patients included one with HIV.[22,23] Thalidomide is known to have a powerful anti-cytokine effect and inhibits tumour necrosis factor and other pro-inflammatory cytokines.
8.10 Anti-Kaposi’s Sarcoma Herpesvirus Therapy
As MCD has been shown to be a viral-driven disease, with the presence of viral genes such as vIL-6 having an effect on pathogenesis, the effect of anti-herpesvirus therapy to reduce the KSHV viral load and alleviate disease has been examined in KSHV-associated diseases in the HIV setting. In HIV-positive patients, KS incidence was reduced when prophylactic ganciclovir or foscarnet were used to prevent cytomegalovirus (CMV) retinitis.[24,25] Furthermore, antiviral treatment, which has led to a clinical improvement, has been shown to reduce KSHV viral load in patients with KS,[26] PEL and haemophagocytic syndrome.[27] In a series of three patients treated with ganciclovir, there was a reduction in the frequency of acute symptoms of MCD for two patients treated with oral and intravenous ganciclovir.[28] For the third patient, who was in the intensive care unit, there was resolution of pulmonary and renal failure with intravenous ganciclovir. All the patients had a reduction in KSHV viral load with the ganciclovir therapy, accompanying the resolution of their symptoms. However, the use of foscarnet and cidofovir antiviral therapy was ineffective in an HIV-negative MCD patient with proven KSHV viraemia and treatment with corticosteroids in combination with chlorambucil chemotherapy was required to achieve a clinical response.[29] Furthermore, the KSHV viral load rose in this patient with the commencement of anti-herpesvirus therapy; this may indicate that the antiviral therapy was ineffective in this case, or that, once the MCD is established, KSHV has a less prominent role and antiviral therapy is less effective than immunotherapy or chemotherapy.
8.11 Rituximab
The use of an anti-CD20 monoclonal antibody, rituximab, routinely prescribed as therapy for B-cell lymphomas and autoimmune diseases, to target KSHV-infected plasmablasts in MCD is a novel and potentially beneficial approach to the treatment of this disease. It has been the subject of case reports in a total of 11 patients. These patients were often pretreated with chemotherapy and follow-up was brief; however, most patients (nine of 11) experienced a complete response.[30-36]
The efficacy and safety of rituximab in 21 consecutive patients with plasmablastic MCD have been investigated.[37] These individuals received four infusions of rituximab 375 mg/m2 at weekly intervals and, of 20 evaluable patients, all achieved clinical remission of symptoms, and biochemical and haematological normalization, and 70% achieved a radiological response. The overall survival and disease-free survival at 2 years were 95 and 79%, respectively, and in three patients who relapsed, retreatment with rituximab was successful. These data corroborate the benefit seen in the aforementioned case reports and demonstrate that rituximab therapy results in an impressive clinical, biochemical and radiological sustained response in HIV-related MCD. Re-treatment with four infusions of rituximab at weekly intervals on relapse has been undertaken in three patients and is safe and effective.[38]
In a further study of 24 patients dependent on chemotherapy for a median time of 13 months, sustained remission was achieved in 70% with this regimen of rituximab and cessation of chemotherapy at day 60 (the primary endpoint).[4] In each of these large series, one patient died soon after rituximab administration as a result of overwhelming disease, and the main adverse event seen in these patients was reactivation of KS, which is intriguing and may have been attributable to the rapid B-cell depletion that is observed during rituximab therapy, or an immune reconstitution inflammatory syndrome to hitherto latent antigens.[39] Rituximab is associated with a decrease in KSHV viral load in the majority of the reports.
8.12 Key Recommendations
|
8.13 References
|
9.0 Other Non-AIDS-defining Malignancies
9.1 Introduction
This section aims to address the evidence-based guidelines for non-AIDS-defining cancers in people with HIV infection. It will exclude Hodgkin’s disease and anal cancer which have been covered already. The cancers it will specifically address are as follows:
|
There are very limited data available on:
|
Therefore these patients should be managed by oncologists and HIV doctors together according to standard guidelines for HIV-negative patients.
9.2 Testicular Germ Cell Cancers
9.2.1 Introduction. It appears that only seminoma (as opposed to nonseminoma) occurs more frequently in HIV-infected patients compared with HIV-negative controls.[1] There is no clear consensus on the exact relative risk but it ranges between approximately 3 and 7.[1-5] There is no evidence that the incidence is increasing in the era of highly active antiretroviral therapy (HAART).[1] The cause for this increased incidence is unclear, although chronic immune suppression has been suggested. Patients present with only moderate immune suppression and they appear to be about 10 years younger than their HIV-negative counterparts.[1] There is conflicting evidence that patients present with more advanced disease. This may be because of the increased incidence of para-aortic lymphadenopathy in HIV disease incorrectly up-staging patients from stage I to stage II disease.[6] Patients with HIV-related testicular cancer have a similar cancer-free outcome compared with their HIV-negative counterparts if treated in an identical manner in the HAART era.[7] This contradicts early reports in the pre-HAART era.[8]
9.2.2 Diagnosis, Staging and Prognostic Factors. Diagnosis should follow an identical path to diagnosis in HIV-negative patients. A testicular mass must be treated with the utmost suspicion and an ultrasound scan or magnetic resonance imaging (MRI) and tests for tumour markers α-fetoprotein (AFP) and human chorionic gonadotrophin (HCG) should follow. Lactate dehydrogenase (LDH) is nonspecific and should only be used to prognosticate patients with metastatic disease.[9] False-positive AFP can be related to HAART/hepatitis-related liver disease, while there are many causes of a false-positive LDH.[1]
The differential diagnosis for a testicular mass in this setting includes orchitis and lymphoma (which occurs in a more elderly population and is associated with a poorer prognosis). A computed tomography (CT) scan of the chest abdomen and pelvis should be performed for full staging. MRI scanning for para-aortic lymph nodes is an alternative for the abdomen and pelvis. There is no clear role for fluorodeoxyglucose positron emission tomography (FDG-PET) in these patients (either HIV-positive or HIV-negative).
9.2.3 Management. 9.2.3.1 Stage I Disease. Patients with stage I disease, either seminomas or nonseminomatous germ cell tumours (NSGCT), can be safely treated with surveillance alone following orchidectomy and have a similar outcome to their HIV-negative counterparts.[7] Alternatively, adjuvant carboplatin (area under the curve × 7 dosing) can be offered to the seminoma patients (we advise one cycle), while two cycles of adjuvant BEP (bleomycin, etoposide and cisplatin) can be offered to the NSGCT patients.[7] It appears that three cycles of BEP suppress the CD4 cell count by between 25 and 50%,[1] and it is probable that two cycles of BEP will also be suppressive. Therefore, low-risk NSGCT patients should be offered surveillance, and adjuvant therapy should only be considered for high-risk NSGCT patients.[6] Additionally, it has been suggested that adjuvant therapy should be considered in patients with a haphazard life-style (who are unlikely to co-operate with an intensive surveillance programme).[6] Patients should continue HAART during adjuvant chemotherapy.[1,7] Prophylactic antifungal agents should be considered, especially for patients receiving two cycles of BEP.[6]
9.2.3.2 Metastatic Disease. Patients should be risk stratified according to the International Germ Cell Cancer Consensus Group (IGCCCG) guidelines in an identical manner to HIV-negative patients.[9] Good-risk patients should be offered three cycles of standard 5-day BEP with concurrent HAART.[6,7] One should expect a 50% drop in the CD4 cell count with chemotherapy[6,10] Treatment modifications should follow the HIV-negative model. Those with extensive pulmonary limitation from previous infection can alternatively have four cycles of EP (etoposide and cisplatin) chemotherapy.[9] Carboplatin should not be used as a substitute for cisplatin and dose modifications/delays should be avoided where possible. Growth factors such as granulocyte colony-stimulating factor (G-CSF) should be considered where appropriate.[9]
Patients with intermediate- and poor-risk disease should be offered four cycles of standard 5-day BEP chemotherapy.[1,6,9] Those with extensive pulmonary limitation from previous infection can alternatively have four cycles of VIP (etoposide, cisplatin and ifosfamide) chemotherapy. The two regimens have similar outcomes in HIV-negative patients but VIP is more myelosuppressive in these patients.[9] Other regimens for poor-risk patients (such as high-dose therapy and dose-dense therapy) have not been shown to be superior to four cycles of BEP in HIV-negative patients and should be used with caution if at all in the HIV-positive population. Patients should receive concurrent HAART and antifungal prophylaxis may be considered where appropriate.
There are very limited data on the treatment of relapsed disease.[1] Patients should be treated in an identical manner to HIV-negative patients. The TIP (paclitaxol, ifosfamide and cisplatin) regimen seems appropriate for patients who relapse 6 months after initial diagnosis.[9] Other regimens such as high-dose therapy should be considered for early relapse. Third-line therapy is usually palliative and there are no data regarding this in HIV-positive patients. It is clear that single-agent therapy has little activity in this setting in HIV-negative patients.
9.2.4 Summary.
|
9.3 Non-small Cell Lung Cancer
9.3.1 Introduction. It appears that the incidence of non-small cell lung cancer (NSCLC) is increasing in people with HIV infection.[11,12] Not all of this increase in incidence can be attributed to smoking cigarettes,[11] although cessation of smoking should be recommended for the HIV-positive population. There is no evidence of an increased incidence of small cell lung cancer in HIV-infected patients and there are no specific data on this issue.[11,12] It is recommended that these patients with small cell lung cancer are treated in an identical manner to their HIV-negative counterparts. What anecdotal data are available suggest that these patients do badly.
Patients with HIV-related NSCLC present at a younger age and with more advanced disease than their HIV-negative counterparts.[11,12] There has been a debate about the increased incidence of adenocarcinomas in the HIV-positive patients. However, studies show that age-matched HIV-negative patients with lung cancer also show an increased incidence of adenocarcinomas.[14]
Studies in the pre-HAART era showed that HIV-positive NSCLC patients have a significantly worse outcome compared with their HIV-negative counterparts. These studies were small and were not all age- or stage-matched; however, the results were compelling with a median survival time of only 3 months.[15,16] Unfortunately, HAART has not had a huge impact on the survival of these patients and the median survival time is still only 4 months.[13] Data suggest that a small minority of these patients are being offered curative surgery.[14] This is attributable to a combination of patients presenting with advanced disease and comorbidity. It is clear that there is a delay in the diagnosis of these patients and this may in part be a result of the wide differential diagnosis of an HIV-infected patient with a mass in the lungs.[14]
9.3.2 Management. As HIV-infected patients with NSCLC present at a younger age than HIV-negative patients, a mass on chest X-ray should raise the suspicion of NSCLC. It is recommend that once a tissue diagnosis has been achieved patients go on to have a CT scan of the chest and abdomen (including adrenals) and a bone scan. If an individual is still potentially operable a mediastinoscopy should be performed. In view of the possible decreased specificity and lack of data regarding FDG-PET in HIV-positive lung cancer its results should be interpreted with caution. Patients should not necessarily be deemed inoperable on the evidence of FDG-PET alone. The results of FDG-PET should be considered in conjunction with their HIV status (HIV history, opportunistic infections, viral load and CD4 cell counts). A CT brain scan is not routinely required in this group.
9.3.2.1 Operable Disease. These patients should be offered surgery, once staging investigations are complete. There are no data regarding the use of adjuvant chemotherapy in HIV-related lung cancer, and therefore management of these patients should follow the HIV-negative lung cancer guidelines. Chemotherapy should consist of standard regimens and doses. HAART should continue throughout treatment. Follow-up should be as with HIV-negative patients.
9.3.2.2 Locally Advanced Disease. There are no data specifically addressing this issue. Patients with locally advanced disease should be offered chemoradiation according to HIV-negative guidelines. It is noteworthy that this treatment has been associated with profound immunosuppression in other HIV-positive tumours.[17] Therefore, patients should continue/commence HAART and antifungal prophylaxis where appropriate.
9.3.2.3 Metastatic Disease. There are data on the management of patients with metastatic disease in the HAART era.[13] They suggest that these patients have a poor outcome, although this may not be different from that of a stage-, histology-, age- and sex-matched HIV-negative cohort (these patients also have a poor outcome). Standard chemotherapy regimens were used for the HIV-positive patients in this study. Unfortunately, treatment was tolerated poorly and response rates were low (<30%). All deaths were attributable to cancer, and no opportunistic infections occurred during chemotherapy. HAART and chemotherapy were given concurrently. As it was conceivable that the HAART increased the chemotherapy toxicity, the authors suggested that interrupting the HAART may be appropriate if the patient’s HIV infection is well controlled.[13,14]
There are currently no data on second- and third-line chemotherapy for metastatic NSCLC. Management should follow HIV-negative guidelines. Tyrosine kinase inhibitors (TKIs) such as erlotinib and gefitinib should be administered with caution as they have not been used in this setting previously and can potentially interact with HAART. This is because they are metabolized by the cytochrome P450 isoenzyme CYP3A4. Data for Kaposi’s sarcoma (KS) suggest that TKIs do indeed potentiate the side effects of HAART.[18]
There are no data on screening this population and no studies are planned. This is partly because of fears regarding the low specificity in this setting. Screening for this population is not recommended with either Chest X Ray (CXR) or CT.
9.3.3 Summary.
|
9.4 Hepatocellular Cancer
9.4.1 Introduction. There is debate as to whether there is an increased incidence of hepatocellular cancer (HCC) in HIV-positive individuals. This uncertainty is primarily because hepatitis B virus (HBV) and hepatitis C virus (HCV) act as confounding factors in this setting. In view of the long delay between development of cirrhosis and subsequent HCC in both HIV-positive and HIV-negative populations, an increase in the incidence of this disease in HIV-infected patients may not have occurred yet.[19]
In Western countries, approximately 30% of people with HIV are coinfected with HCV, rising to approximately 75% in injecting drug users.[20] HIV affects the natural history of HCV infection in two important ways: firstly, it increases the likelihood of chronic infection following the acute episode and, secondly, it hastens the development of cirrhosis once chronic infection is established. This has important implications for the subsequent development of HCC and screening strategy.[19]
HIV coinfection also accelerates the progression of HBV infection.[21] There is a large regional variation in the proportion of people with HIV who have previously been exposed to HBV (10-90%). Retrospective series suggest that HBV is responsible for a much smaller proportion of HCC compared with HCV in HIV-positive individuals.[19,20]
9.4.2 Presentation and Diagnosis. The largest study to compare HIV-positive and HIV-negative patients with HCC consisted of 41 cases diagnosed between 1989 and 2002.[20] The majority of the HIV-infected cohort had HCV and cirrhosis. The HIV-positive patients were only moderately immune-suppressed at presentation. Seventy-seven per cent were on HAART at diagnosis and only 24% had a previous AIDS-defining illness. The HIV-infected patients were younger and more often HCV-positive, and presented with more advanced infiltrating cancers compared with HIV-negative patients. Fewer of the HIV-infected patients were offered active treatment and survival rates were poor. However, when patients were offered interventional treatment such as resection or transplantation their outcome was similar to that of the HIV-negative population.[19,24-26]
Most HCCs are identified with ultrasound (US) scanning and measurement of AFP levels.[20] The degree of cirrhosis should be assessed prior to any definitive treatment using the Child-Pugh classification. If complete resection is possible this should be performed without biopsy. These patients should have category A cirrhosis according to the Child-Pugh classification.[22] Therefore, a CT scan of the chest, abdomen and pelvis is required to exclude metastatic disease. Data exist on liver transplantation in this setting and this should be offered to HIV-infected patients if appropriate[23] according to the Milan criteria and includes three lesions <3cm or one lesion <5 cm in diameter. Once again biopsy should be avoided. It appears that transplantation may have superior results to resection alone in HIV-negative patients [University of California San Francisco (UCSF) criteria].[27]
9.4.3 Management. In the HIV-negative population, solitary or a small number of HCC lesions are resectable, and associated with a 5-year survival of 60-70%.[28] In the presence of cirrhosis, patients with operable lesions are offered transplantation resulting in equivalent survival data.[27] Ethanol injections are another treatment option for patients with local disease which is associated with 5-year survival rates of approximately 50%. Patients with more advanced disease are limited to palliative embolization. No chemotherapy or targeted therapy has been shown to offer a survival benefit for these patients.
Published series in HIV-positive individuals with HCC show that the majority (60%) of HIV-positive HCC patients are not being offered active treatment, because of the advanced nature of their disease.[20] They have a worse outcome compared with their HIV-negative counterparts, with only 28% 1-year survival. However, further analysis shows that when HIV-infected patients are offered active treatment they have a similar survival to their HIV-negative counterparts.[24-26] This treatment included both liver resection and transplant, although published data are rather limited for the former.
Special attention is required for HIV-positive liver transplants because of the potential interaction between HAART and immunosuppressive therapy such as tacrolimus. This is particularly true for inhibitors of cytochrome P450 such as protease inhibitors.
9.4.4 Screening for HCC in Patients with Hepatitis and HIV Coinfection. The European Association for Study of the Liver (EASL) has released guidelines for screening HIV and HCV/HBV coinfected individuals. They recommend that HBV/HIV coinfected patients be screened for HCC every 6 months with ultrasonography and measurement of AFP levels. The American Gastroenterological Association (AGA) feels that AFP is unreliable because of the very high false-positive rate. None of these data has been validated. The UK group feels that 6-monthly US scans for HIV-positive HCV-infected patients with cirrhosis are advisable. Because HBV is potentially oncogenic, in the absence of cirrhosis we still advise that all these coinfected patients have 6-monthly US scans. The role of AFP in this setting is unknown and no consensus can therefore be reached. Studies are required to justify this screening programme.
9.4.5 Summary.
|
9.5 Other Cancers
Only small retrospective series in the pre-HAART era exist for other malignancies. There are no data comparing these cancers in HIV-infected patients with those in HIV-negative patients, and no data in the HAART era. However, there are anecdotal data to suggest that head and neck cancers, colon cancers and breast cancers may be more aggressive than in their HIV-negative counterparts. One of the more intriguing issues regarding these tumours is the decreased incidence of prostate and breast cancer in HIV-infected patients.[2,29] The reason for this decrease does not appear to be related to hormone deficiency.[29] Further work is required. We recommend that patients with these less well-described cancers are offered the standard care offered to HIV-negative patients. Treatment should be given in consultation with HIV doctors. Prospective databases are required for this group.
9.5.1 Summary.
|
9.6 References
|
10.0 Highly Active Antiretroviral Therapy (HAART) and Prophylaxis of Opportunistic Infections
10.1 Introduction
HIV infection is associated with immunosuppression, CD4 lymphocyte count loss and a progressive risk of opportunistic infection and tumours. Chemotherapy for HIV-related malignancies is associated with an increased risk of infection secondary to the myelosuppression associated with chemotherapy, additional CD4 lymphocyte count loss with chemotherapy for both Kaposi’s sarcoma and lymphoma,[1] the presence of central venous catheters[2-5] and neutropenia associated with HIV,[6,7] and many of the therapies utilized to treat HIV infection and its complications.[8-10]
Guidelines for the initiation of opportunistic infection prophylaxis[11] and highly active antiretroviral therapy (HAART) are available.[12,13] However, there may be other considerations for earlier instigation of HAART and opportunistic infection prophylaxis in those receiving chemotherapy for HIV-associated malignancies. As non-Hodgkin’s lymphoma (NHL), primary cerebral lymphoma (PCL) and Kaposi’s sarcoma (KS) are AIDS-defining events, all individuals receiving these diagnoses should commence HAART unless otherwise indicated. Although guidelines suggest initiation of prophylaxis against opportunistic infections based on CD4 cell count,[11] this does not apply in those with malignancies because of the possible profound immunosuppression associated with chemotherapy.
Prophylaxis against Pneumocystis jirovecii pneumonia (PCP) is recommended for those who have a CD4 count <200 cells/µL and should be considered in individuals with an AIDS diagnosis with a CD4 cell count above this level.[11] Individuals diagnosed with NHL, PCL or KS should therefore always be considered for PCP prophylaxis and consideration should be given to initiating prophylaxis in other individuals who receive immunosuppressant chemotherapy for other tumours. Mycobacterium avium complex (MAC) prophylaxis is recommended for HIV-positive individuals with a CD4 count below 50 cells/µL but may be considered in all patients receiving chemotherapy associated with immunosuppression.
Primary prophylaxis against fungal infections is not recommended because of a lack of data on efficacy and the risk of development of infection with resistant organisms. However, individuals who have prolonged central venous access via either a Hickman line or a Portacath should be considered for antifungal prophylaxis where there is a high risk of development of invasive fungal infection.
10.2 HAART
There is a wide choice of HAART available, and the British HIV Association (BHIVA) Treatment Guidelines should be followed.[13]
In antiretroviral (ARV)-naïve individuals, treatment regimens should consist of two nucleoside reverse transcriptase inhibitors (NRTIs) in combination with either a nonnucleoside reverse transcriptase inhibitor (NNRTI) or a ritonavir-boosted protease inhibitor (PI). There appears to be no difference in response of KS to NNRTI- and PI-based therapies.[14] There are no data on choice of NNRTI or PI for lymphoma. Ritonavir (RTV) is a potent inhibitor of the P450 microsomal enzyme system and also inhibits glyco-protein efflux pumps which may lead to increased toxicity to some chemotherapeutic agents. The use of an RTV-boosted PI has been associated with more profound and prolonged neutropenia when used with therapy for NHL[15] and NNRTI is the treatment of choice unless there are contraindications.
In individuals who develop malignancies receiving a nonsuppressive ARV regimen, strong consideration should be given to changing the therapy to ensure full viral suppression and the best immunological activation possible.
10.2.1 Nucleoside Reverse Transcriptase Inhibitors. Zidovudine (ZDV) – avoid because of increased risk of anaemia and myelosuppression.[8]
Stavudine (d4 T) – not recommended in present treatment guidelines;[13] possible increased risk of peripheral neuropathy with neurotoxic chemotherapy.
Didanosine (ddI) – avoid in individuals receiving neurotoxic chemotherapy.
Tenofovir (TDF) – avoid in individuals receiving reno-toxic chemotherapy.
Abacavir (ABC) – no contraindication.
Lamivudine (3TC) – no contraindication.
Emtricitabine (ETC) – no contraindication.
10.2.2 Nonnucleoside Reverse Transcriptase Inhibitors. Efavirenz (EFV) – no increased risk of toxicity.
Nevirapine (NVP) – avoid in individuals receiving hepatotoxic chemotherapy.
10.2.3 Protease Inhibitors. Avoid RTV-boosted PIs if there is a risk of interaction with chemotherapy.
Saquinavir – reported to be associated with a higher risk of mucositis when given with cyclophosphamide, doxorubricin, eroposide (CDE).[16]
Unboosted PIs – not recommended for HIV therapy.[12,13]
In individuals initiating HAART after the development of malignancy, recommended regimens are either Truvada (tenofovir/emtricitabine) and efavirenz or Kivexa (abacavir/lamivudine) and efavirenz.
10.3 Opportunistic Infection Prophylaxis
10.3.1 Bacterial Infections. Individuals receiving chemotherapy are at risk of developing neutropenia. The risk of neutropenia is increased both by agents utilized to treat HIV infection and its complications and by HIV infection itself.[6-10]
Several studies have shown an increased risk of infectious complications in neutropenic HIV-positive individuals.[5,17,18] Where this is prolonged, or in the presence of a central venous catheter, the frequency of bacterial infection may be reduced by the use of prophylactic granulocyte colony-stimulating factor (G-CSF) to reduce the trough neutrophil count and the duration of neutropenia.[19,20]
Individuals should avoid other myelosuppressive agents while receiving chemotherapy, particularly ZDV and ganciclovir. Pre-HAART, G-CSF was possibly associated with increases in HIV viral load;[21,22] however, this should not be an issue in individuals receiving fully suppressive HAART.
Untunnelled central venous catheters may be associated with lower infection rates[23] as may peripherally inserted central catheters.[24] The commonest organisms associated with this complication are Staphylococcus aureus, including methicullin resistant S. aureus (MRSA), which may be linked to the high rates of skin and nasal infection in HIV-positive individuals, and coagulase-negative staphylococci which are likely to be associated with breaches of infection control.[25] Antibiotic lock procedures may reduce infection rates of catheters although data are lacking in the context of HIV.[26]
The issue of antibiotic prophylaxis to reduce the incidence of life-threatening bacterial infection in chemotherapy-induced neutropenia has recently come to the fore. The benefits of antibiotic prophylaxis in reducing infection in neutropenic patients is well documented, with a meta-analysis evaluating trials using fluoroquinolones demonstrating a significant reduction in gram-negative infections, microbiologically documented infections, total infections and the incidence of fever when compared with no prophylaxis.[27]
A meta-analysis of the addition of gram-positive cover to fluoroquinolone prophylaxis demonstrated reductions in bacteraemia and both streptococcal and staphylococcal infections but no association with improvement in mortality secondary to infection.[28]
More recently a randomized study of ciprofloxacin and roxithromycin or placebo in small cell lung cancer patients treated with CDE reported a reduction in the number of infectious deaths from 6 to 0%.[29]
A meta-analysis of 95 randomized trials of nearly 10 000 patients has recently been published. Fifty of these trials compared antibiotic to placebo and used 45 different anti-prophylactic regimes. Fifty-two of the trials addressed the use of fluoroquinolone prophylaxis. Overall antibiotic prophylaxis was associated with a reduction in overall mortality of 33% and a reduction in mortality associated with infection of 42%; the effect was most marked with fluoroquinolone prophylaxis with reductions of 48 and 62%, respectively.[30]
The SIGNIFICANT trial was a multicentre, randomized, double-blind placebo-controlled study examining the use of levofloxacin in individuals receiving cyclic chemotherapy for solid tumours and lymphoma at risk of severe neutropenia. Prophylaxis with levofloxacin 500 mg once daily or placebo was given for 7 days to cover the anticipated nadir in neutrophil count. Use of levofloxacin was associated with a reduction of over 50% in the instance of fever during chemotherapy, with protection being most marked during the first cycle of chemotherapy. Probable infections were reduced by 29% and hospitalization by 36%.[31]
The GIMEMA study examined levofloxacin prophylaxis in patients at high risk of neutropenia (those with acute leukaemia or undergoing high-dose chemotherapy) and also reported improvements in infectious complications but not mortality.[32]
Clinicians considering the use of prophylactic antibiotics to prevent neutropenic sepsis must also consider the possible emergence of resistant micro-organisms.
There are no data on the use of antibiotic prophylaxis for the prevention of bacterial infections in the HIV setting. The use of cotrimoxazole in HIV-infected individuals to prevent PCP may give some protection against bacterial infections although studies confirming reduction in catheter-related infections are lacking.[33-35]
10.3.2 Antifungal Prophylaxis. Individuals infected with HIV with low CD4 cell counts are at risk of fungal infections, most commonly oral and oesophageal candida and cryptococcosis, while those with prolonged very low CD4 cell counts are also at risk of pulmonary aspergillosis. In individuals with central venous catheters in situ and profound neutropenia, invasive fungal infections are a considerable cause of morbidity and mortality.
Because of the CD4 decline associated with chemotherapy, neutropenia and the necessity in some individuals to receive chemotherapy via a central venous catheter, the use of azole prophylaxis should be considered. Although primary prophylaxis against oro-oesophageal candida and cryptococcosis is not recommended because of the risk of development of resistance,[11,36] the use of prophylactic azoles may reduce the incidence of these conditions.
The choice of azole is between itraconazole and fluconazole. In individuals with haematological malignancies and neutropenia, a recent meta-analysis of fluconazole vs. itraconazole as fungal prophylaxis revealed no statistically significant differences in documented invasive fungal infections, overall mortality and mortality attributed to fungal infection. However, prophylactic use of fluconazole resulted in significantly more fungal infections when documented and suspected infections were combined. Conversely, fewer individuals developed treatment-limiting toxicities with fluconazole.[37] There are potential drug interactions between vinca alkaloids and itraconazole, so fluconazole should be used with chemotherapy regimens including vincristine, vinblastine, vindesine and vinorelbine.[38]
A more recent publication compared oral itraconazole solution with fluconazole oral solution for primary prophylaxis of fungal infections in individuals with haematological malignancy and profound neutropenia. There were no statistical differences in the efficacy or safety of itraconazole and fluconazole.[39]
Newer antifungal agents such as posaconazole and voriconazole may have advantages over existing agents with regard to efficacy of prophylaxis, although the cost of such agents may be prohibitive and there are potential drug interactions.[40-42]
10.3.3 Pneumocystis jirovecii pneumonia prophylaxis. All individuals with a CD4 count below 200 cells/µL should receive prophylaxis against PCP, and this should be considered for all individuals with an AIDS diagnosis with a CD4 cell count above this level.[11] All patients developing a malignancy should be considered for commencement of PCP prophylaxis if they are receiving chemotherapy or radiotherapy.
The treatment of choice is cotrimoxazole, which may have additional benefits in reducing the incidence of bacterial infections [respiratory, gastrointestinal (especially salmonella) and possibly central nervous system infections][33-35,43] and toxoplasmosis.[44,45] Alternative prophylaxis should be with dapsone or pentamidine via nebulizer.
10.3.4 Mycobacterium avium complex prophylaxis. Prophylaxis against MAC is recommended for individuals with a CD4 count <50 cells/µL.[11] Individuals who have or are at risk of a CD4 cell count falling below this level should be considered for MAC prophylaxis. The treatment of choice is azithromycin 1.25 g once per week or clarithromycin with rifabutin being considered as an alternative.[46-49]
10.3.5 Hepatitis B. It is estimated that over 50% of individuals with HIV have markers of previous hepatitis B virus (HBV) infection, with (in one pan-European study) 8.7% having active hepatitis B, defined as being surface antigen-positive.[50] This percentage may be even greater in some racial groups where there is a higher natural prevalence of hepatitis B. Occult hepatitis B (viral replication in the absence of surface antigen) is well documented in those with HIV[51,52] and reactivation of HBV has been documented at low CD4 cell counts.[53,54] The risk of reactivation of HBV and increases in HBV DNA with immunosuppressive chemotherapy has been documented in both the HIV-negative and HIV-positive settings.[55-59]
Li et al. recently reported on the use of prophylactic lamivudine in individuals receiving chemotherapy for lymphoma, comparing 40 patients who received lamivudine with 116 historical controls who did not. The use of lamivudine was associated with a lower incidence of severe hepatitis and fewer disruptions of chemotherapy. Overall mortality in those receiving lamivudine was lower compared with the historical controls, although this did not reach statistical significance.[60]
Rossi et al.[61] have reported a pilot study to assess the use of lamivudine in individuals who were HBV surface antigen-positive. No signs of HBV activation developed. Another study reported the use of lamivudine or interferon in HBV surface antigen-positive individuals receiving chemotherapy for lymphoma. The use of lamivudine or interferon was associated with a reduction in grade 3 or 4 toxicity-elevated alanine aminotransferase (ALT). Thirty-two per cent of the nonprophylactic group experienced HBV reactivation, 41% of whom progressed to fulminant hepatitis.[62]
In individuals with actively replicating HBV commencing HAART, therapy should include drugs active against HBV, i.e. tenofovir with either lamivudine or emtricitabine.[63] Individuals with replicating HBV or who are at risk of HBV activation and not requiring HAART should receive prophylaxis/treatment with entecavir or adefovir.
10.4 Recommendations
Kaposi’s sarcoma
|
NHL/PCL
|
Other malignancies
|
Hepatitis B
|
Bacterial infection prophylaxis
There is insufficient evidence to allow recommendations to be made on the use of antibacterial prophylaxis to prevent central venous catheter-related bacterial sepsis. When considering the use of such prophylaxis, the positive results in HIV-negative studies should be balanced against the risk of development of resistance and the potential extra cost involved.
10.5 References
|
CLICK HERE for subscription information about this journal.
Table 1. Evidence Levels Used in Guidelines

Table 2. The Modified AIDS Clinical Trials Group Staging of Kaposi’s Sarcoma (KS)[1,2]
![Table 2: The Modified AIDS Clinical Trials Group Staging of Kaposi's Sarcoma (KS) [1,2]](http://images.medscape.com/images/583/877/art-hiv583877.tab2.gif)
Table 3. Response Criteria for HIV-associated Kaposi’s Sarcoma (KS)[1]
![Table 3: Response Criteria for HIV-associated Kaposi's Sarcoma (KS) [1]](http://images.medscape.com/images/583/877/art-hiv583877.tab3.gif)
Table 4. The Results of Phase III Trials of Liposomal Anthracyclines for Kaposi’s Sarcoma (KS)
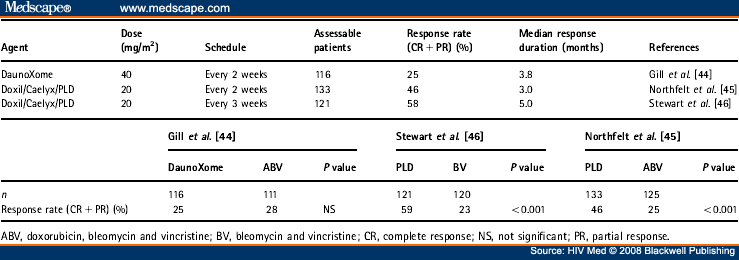
Table 5. Baseline Investigations*

Table 6. Ann Arbor Classification/Cotswolds Modification

Table 7. International Prognostic Index (IPI) for Aggressive Non-Hodgkin’s Lymphoma

Table 8. International Federation of Gynecology and Obstetrics (FIGO) Staging of Cervical Cancer

Table 9. TNM Definitions

Table 10. Baseline Investigations in HIV-associated Hodgkin’s Lymphoma

Table 11. Ann Arbor Classification/Cotswolds Modification for Staging HIV-associated Hodgkin’s Lymphoma

Table 12. International Prognostic Score (IPS) Hasenclever Index for Hodgkin’s Lymphoma

Table 13. Published Studies of First-line Therapy in HIV-associated Hodgkin’s Lymphoma
Table 14. Published Studies of Therapy in Relapsed HIV-associated Hodgkin’s Lymphoma (HL)
![]()
Dr Mark Bower, Department of Oncology, Chelsea & Westminster Hospital, 369 Fulham Road, London SW 10 9NH, UK. Tel: 0208 237 5054; fax: 0208 746 8863; e-mail: m.bower@imperial.ac.uk
![]()
1Department of Oncology, Chelsea & Westminster Hospital, London, UK
2HIV i-Base and UK-CAB, London, UK
3St Bartholomew’s Hospital, London, UK
4Royal Free Hospital, London, UK
5Hammersmith Hospital, London, UK
6Royal Sussex County Hospital, Brighton, UK
Tags: News, Rianimazione




Leave a Reply
You must be logged in to post a comment.