Thrombosis is a major cause of morbidity and mortality worldwide. Traditional anticoagulant therapies (heparin and warfarin), although efficacious, are associated with limitations in their utility for long-term administration. Consequently, there is an ongoing search for new anticoagulant agents. A better understanding of the biochemical pathways involved in coagulation has led to the identification of new targets for anticoagulant agents. This article reviews the key events in the cell-based model of coagulation, the rationale for current and new targets for anticoagulant agents, and the efficacy and safety of new oral anticoagulant agents in late-stage clinical development.
Introduction
Coagulation is the process through which blood clots are formed at sites of injury to blood vessels. Such clots must form rapidly to arrest hemorrhage from the wound, but once formed, the clotting process must be quickly regulated to prevent the clot from expanding to the point where it obstructs blood flow. Once bleeding is controlled, the fibrinolytic system degrades the blood clot so that blood flow is restored and healing can occur.
Coagulation involves a regulated sequence of events in which a series of inactive enzyme precursors, or zymogens, become activated. The end result of this process is the timely generation of thrombin, which is also known as factor IIa (FIIa), the enzyme that not only converts fibrinogen to fibrin but also amplifies its own generation and serves as a potent platelet agonist. The original “cascade” or “waterfall” models of coagulation proposed 2 independent pathways by which coagulation could be initiated in a platelet-free environment.[1,2] Although the concept of distinct extrinsic and intrinsic pathways is useful for understanding in vitro tests of coagulation, we now know that these pathways are intricately linked. We also know that platelets and other cellular components of the blood are integral parts of the coagulation system. Building on this information, a cell-based model of coagulation has evolved in an attempt to better understand the complex reactions that occur on the surface of cells and the integration of processes that result in the formation of a platelet-fibrin thrombus.[3-5]
According to the cell-based model of coagulation, the initiating or triggering event occurs on the surface of intact tissue factor (TF)-bearing cells or cell fragments, known as microparticles, which are exposed or generated at sites of vascular injury.[4,5] Such cells include monocytes, macrophages, and smooth muscle cells among others. As a transmembrane protein, TF on the surface of these cells binds circulating FVIIa and the resultant TF:FVIIa complex initiates the generation of a small amount of thrombin (the spark). This thrombin then amplifies its own generation, which results in a burst of thrombin (the flame) that converts fibrinogen to fibrin, stabilizes the fibrin network, and activates platelets.[4,5] The precisely synchronized sequence of events is counterbalanced by a system of anticoagulant and fibrinolytic mechanisms, which serve to ensure that the hemostatic effect is regulated and does not extend inappropriately. In pathological states, these events can escape normal control mechanisms due to either inherited or acquired defects, resulting in thrombosis, which is the generation of an occlusive thrombus that obstructs or limits blood flow in an artery or vein.
Based on current knowledge of the main triggers and a better understanding of the structure and function of the key coagulation enzymes, anticoagulant therapy is moving towards targeted therapy. This article reviews our current understanding of the coagulation system, identifies the major enzymes in this pathway, and provides a rationale for the selection of targets for novel anticoagulants.
Triggers of Coagulation
Injury to the vessel wall is considered to be the primary stimulus that sets coagulation in motion. Vessel injury may be mechanical, chemical, or electrical. In arteries, vessel injury is mainly due to the rupture of an atherosclerotic plaque, resulting in disruption of the endothelium and exposure of plaque constituents to the blood (Figure 1).[6] In veins, vessel wall disruption is a less important trigger of coagulation, although vessel wall injury can occur in the setting of surgery or with indwelling central venous catheters. More common triggers are reduced blood flow (stasis), altered properties of the blood that render it more prone to clotting (thrombophilia), and/or alterations in the endothelium.[6]

Figure 1. Triggers of arterial thrombosis and venous thrombosis. A = artery; b = vein. From Mackman N. Nature. 2008;451:914-918.6 Reprinted with permission.
The endothelium contains at least 3 thromboregulators — nitric oxide,[7,8] prostacyclin,[9] and the ectonucleotidase, CD39[10] — that are responsible for maintaining an anticoagulant milieu and providing a defense against thrombus formation in an uninjured vessel. In addition, subendothelial collagen and TF facilitate the maintenance of a closed circulatory system. Vessel disruption exposes subendothelial collagen and TF to the circulating blood. Exposed collagen triggers the accumulation and activation of platelets, whereas exposed TF is the major initiator of coagulation.[6]
Initiation of Coagulation
TF, also known as FIII, is a cell-bound transmembrane glycoprotein that is constitutively expressed by fibroblasts and smooth muscle cells of the vessel wall.[5] It is also constitutively expressed in many tissues such as the brain, heart, lungs, kidneys, testes, and placenta, highlighting the importance of this glycoprotein.[11] Inflammatory stimuli (eg, bacterial lipopolysaccharide in sepsis), adhesion molecules (eg, P-selectin expressed on activated platelets and CD40 ligand expressed on activated white blood cells), inflammatory cytokines (eg, interleukin-6 and tumor necrosis factor), and oxidized low-density lipoprotein(LDL) can induce TF expression in monocytes and endothelial cells.[12] Circulating microparticles derived from monocytes and other cell types have also been shown to express TF on their surface.[13] TF not only plays an important role in coagulation but also mediates intracellular signaling events in inflammation, apoptosis, embryonic development, and cell migration.[14]
TF on the surface of intact and apoptotic cells in the core of atheromatous plaques in arteries is thought to be the main stimulus of arterial thrombosis after plaque rupture.[6] In veins, endothelial cell activation results in the expression of adhesion molecules that then tether TF-bearing monocytes or microparticles onto their surface. It is these tethered cells that are thought to be the stimulus for venous thrombosis.[5] In either case, TF-bearing cells or microparticles provide the key cellular surface for the initiation of coagulation.[5]
Coagulation is initiated when TF binds to circulating FVIIa, forming a catalytic complex, TF-FVIIa, the so-called extrinsic tenase complex, on the phospholipid surface of the TF-bearing cell membrane[4,5] (Figure 2). The extrinsic tenase complex then activates FIX and FX, and the resultant FXa converts a small amount of prothrombin (FII) to thrombin. This small amount of thrombin triggers the amplification phase by activating platelets as well as FV, FVIII, and FXI. Activated platelets provide a surface on which the coagulation factors assemble. FXIa generates additional FIXa, which then binds to FVIIIa on the surface of activated platelets to form the intrinsic tenase complex. This complex propagates coagulation by efficiently activating FX. FXa binds to FVa on the surface of activated platelets to form the prothrombinase complex. A potent activator of prothrombin, the prothrombinase complex triggers a burst of thrombin generation.[4,5]

Figure 2. Overview of the coagulation cascade.
Amplification of Coagulation
The amplification phase of coagulation occurs mainly on platelets, although it may also occur on microparticles, activated endothelium, and other cells. The small amount of thrombin generated during the initiation phase activates platelets and converts FV to FVa, FVIII to FVIIIa, and FXI to FXIa[4] (Figure 2). FIXa, produced in the initiation phase, in the presence of its activated cofactor, FVIIIa, forms the intrinsic tenase complex (FIXa-FVIIIa)[15] on activated platelet surface in the presence of calcium. The intrinsic tenase complex amplifies the activation of FX. FXa, with its activated cofactor, FVa, forms the prothrombinase complex (FXa-FVa) on the platelet surface. The intrinsic tenase complex (FIXa-FVIIIa) and the prothrombinase complex (FXa-FVa) co-localized on the platelet membrane, in the presence of calcium, create a positive feedback loop, the result of which is the rapid generation of thrombin.[16]
Propagation of Coagulation
In the propagation phase, activated platelets are recruited (thrombin is important for this recruitment) to the site of injury to provide appropriate localization of the intrinsic tenase complex, the prothrombinase complex, and calcium on the platelet surface for optimum generation of thrombin. The resulting “thrombin burst” leads to the generation of fibrin from fibrinogen. The soluble fibrin monomers coalesce into a fibrin polymer gel and FXIIIa, activated by thrombin, covalently crosslinks the fibrin strands to form a stable fibrin network[16] (Figure 2).
After clot formation, thrombin generation continues. This additional thrombin not only solidifies the clot but also has other proinflammatory and proliferative functions that promote wound healing. The thrombin formed also activates thrombin-activatable fibrinolysis inhibitor (TAFI), which serves to protect the clot from premature plasmin-mediated fibrinolysis. TAFIa attenuates fibrinolysis by cleaving carboxy-terminus lysine residues from fibrin; this downregulates fibrinolysis because lysine residues, which are exposed on degrading fibrin, represent binding sites for plasminogen and plasmin.[17]
Regulation of Coagulation
Regulation of the coagulation pathway occurs at each phase either by enzymatic inhibition or modulation of cofactor activity.
Tissue factor pathway inhibitor (TFPI), a Kunitz-type inhibitor, regulates the extrinsic tenase complex. TFPI binds to FXa and neutralizes its activity. The TFPI-FXa complex then inhibits FVIIa bound to TF. TFPI is predominantly synthesized by endothelial cells and to a lesser extent by megakaryocytes. There are 4 pools of TFPI in the circulation: a plasma pool of which about 80% is bound to LDL, while the remainder is free; an endothelium-bound pool, which is released after heparin administration; and a small pool within platelets.[18]
Antithrombin, a serine protease inhibitor (serpin), is the most important inhibitor of coagulation. Antithrombin inactivates thrombin, FXa, and other clotting enzymes in a reaction that is accelerated by heparin.[19] In vivo, vessel wall heparan sulfate may substitute for medicinal heparin as the antithrombin activator.
Thrombomodulin, expressed on the surface of endothelial cells, serves as a receptor for thrombin. Once bound to thrombomodulin, thrombin no longer serves as a procoagulant and cannot activate platelets, convert fibrinogen to fibrin, or amplify its generation. Endothelial protein C receptor, also expressed on the surface of endothelial cells, binds to protein C, a vitamin K-dependent protein, and presents it to the thrombin-thrombomodulin complex for activation. Activated protein C, in the presence of protein S as cofactor, degrades FVa and FVIIIa, blocking the amplification of the coagulation system, thus limiting further thrombin formation.[20]
Protein Z, another vitamin K-dependent protein, inhibits FXa via protein Z-dependent protease inhibitor.[21]
Fibrinolysis
Fibrinolysis is essential for degrading a clot once it has served its hemostatic function. Plasmin, the principal mediator of fibrinolysis, degrades fibrin resulting in the production of soluble fibrin degradation products[22] (Figure 3). Plasmin is produced by proteolytic cleavage of circulating plasminogen by plasminogen activators. There are 2 functionally distinct plasminogen activators: tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA). t-PA is released from endothelial cells in response to thrombin and venous occlusion. u-PA is produced as an inactive form, prourokinase, which is activated by plasmin, kallikrein, and FXIIa (the so-called contact factors). Fibrinolysis is regulated at 2 levels. Plasmin is inhibited by α2-antiplasmin, whereas plasminogen activator inhibitors regulate the activity of t-PA and u-PA.[22]

Figure 3. Fibrinolysis
Anticoagulant Therapy
Anticoagulant therapy can be used to prevent or treat arterial or venous thrombosis. Thrombosis occurs when the regulatory mechanisms of hemostasis are overwhelmed by pathologic processes and an excessive amount of thrombin is generated. Arterial thrombosis superimposed on disrupted atherosclerotic plaque is the critical event underlying most cases of myocardial infarction (MI) and stroke.[6] Venous thromboembolism (VTE) occurs in 10%-40% of patients immobilized because of a medical illness or undergoing general surgery, and in 40%-60% of patients undergoing major orthopaedic surgery.[23] VTE, which includes deep vein thrombosis (DVT) and pulmonary embolism (PE), is associated with considerable morbidity and mortality. PE is the main preventable cause of death in hospitalized medical patients and in those undergoing major orthopaedic surgery or extensive surgery for cancer.[23]
Both parenteral and oral formulations of anticoagulant therapy are available. Until recently, the heparins and vitamin K antagonists (VKAs) were the 2 main classes of parenteral and oral anticoagulants, respectively. Although efficacious, these agents are associated with limitations. For example, even though the risk for heparin-induced thrombocytopenia is lower with low-molecular-weight heparin than with unfractionated heparin and almost nonexistent with fondaparinux,[6] all of these agents require subcutaneous injection at least once daily, which limits their utility for long-term administration.
VKAs, such as warfarin, can be given orally, which renders them more acceptable for long-term use. However, these agents have a delayed onset of action because they exert their anticoagulant effect by inhibiting the synthesis of functional vitamin K-dependent coagulation factors (FII, FVII, FIX, and FX), a process which takes several days to occur.[24,25] Warfarin is the most commonly prescribed anticoagulant in the United States.[26,27] Its oral route of administration is its most attractive attribute, but safety concerns remain a substantial issue with its use. Because of warfarin’s narrow therapeutic window and large interpatient variability in dose response, regular monitoring of the international normalized ratio (INR) is essential to ensure that a therapeutic level of anticoagulation is maintained.[26,27] The delayed onset of warfarin action and dose-response variability often prolong hospitalization and increase healthcare costs.[28] Warfarin has numerous interactions with both dietary vitamin K as well as other medications, which contribute to its dose-response variability and necessitate patient counseling before hospital discharge.[26,27]
Newer Oral Anticoagulant Agents
The limitations of warfarin prompted the search for new oral anticoagulants that can be given in fixed doses without the need for routine coagulation monitoring. The agents in most advanced stages of development target either thrombin or FXa (Figure 4). Two such agents, dabigatran etexilate and rivaroxaban, have already been licensed in Europe and Canada (but not currently in the United States) for thromboprophylaxis after elective hip or knee replacement surgery. A third agent, apixaban, is under investigation for this indication.
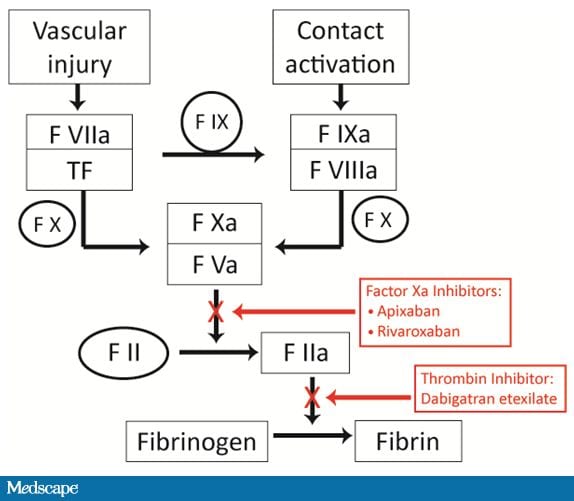
Figure 4. New anticoagulants and their site of action.
These new oral anticoagulants are more specific than warfarin as they target a single clotting enzyme, either thrombin or FXa, and produce such a predictable anticoagulant response because they have minimal drug or food interactions that monitoring is not required (Table 1).[29]
Table 1. Pharmacologic Features of Dabigatran Etexilate, Rivaroxaban, and Apixaban
| Dabigatran Etexilate | Apixaban | Rivaroxaban | |
| Target | Thrombin | Factor Xa | Factor Xa |
| Prodrug | Yes | No | No |
| Dosing | Fixed, once daily | Fixed, twice daily | Fixed, once daily |
| Bioavailability (%) | 6 | 50 | 80 |
| Monitoring | No | No | No |
| Half-life (h) | 12-14* | 12.7 | 7-11 |
| Renal clearance (%) | 80 | 25 | 65 |
| Interactions | P-gp inhibitors** | Potent CYP3A4 inhibitors† | Combined P-gp inhibitors + CYP3A4 inhibitors† |
| *In healthy volunteers, 14-17 hours in patients undergoing major orthopaedic surgery. **P-glycoprotein (P-gp) inhibitors include verapamil, clarithromycin, and quinidine. Quinidine is contraindicated in patients receiving dabigatran. †Cytochrome P450 (CYP) 3A4 inhibitors include ketoconazole, macrolide antibiotics, and protease inhibitors. |
|||
From Gross PL, et al. Clin Pharmacol Ther. 2009;86:139-146.[29]
Dabigatran Etexilate
Dabigatran etexilate is a direct, specific, and reversible thrombin inhibitor (Table 1). It is a prodrug of dabigatran and is rapidly and completely converted to its active form after oral administration.[30]
Three large, phase 3 trials — RE-NOVATE,[31] RE-MOBILIZE,[32] and RE-MODEL[33] — investigated the efficacy and safety of oral dabigatran etexilate vs enoxaparin for the prevention of VTE after major orthopaedic surgery. In RE-NOVATE and RE-MODEL, oral dabigatran etexilate 150 and 220 mg administered once daily was compared to subcutaneous enoxaparin 40 mg once daily in patients undergoing total hip replacement (THR) and total knee replacement (TKR), respectively. In RE-MOBILIZE, dabigatran etexilate 150 and 220 mg was compared with enoxaparin 30 mg twice daily in patients undergoing TKR. In all 3 trials, the primary efficacy endpoint was the composite of total VTE (symptomatic or asymptomatic) and all-cause mortality.
In RE-NOVATE and RE-MODEL, both doses of dabigatran etexilate were noninferior to enoxaparin, though in RE-MOBILIZE, both doses of dabigatran etexilate were inferior to enoxaparin in preventing total VTE and all-cause mortality. The incidence of major bleeding was similar across the 3 treatment groups in all 3 studies (Table 2).
Table 2. Efficacy and Safety Outcomes From RE-NOVATE, RE-MOBILIZE, and RE-MODEL
| Enoxaparin | Dabigatran Etexilate 150 mg |
Dabigatran Etexilate 220 mg |
|||
| DVT, PE, and all-cause mortality (%) | P | P | |||
| RE-NOVATE (THR) | 6.7 | 8.6 | <.0001* | 6.0 | <.0001* |
| RE-MOBILIZE (TKR) | 25.3 | 33.7 | .0009† | 31.1 | .02† |
| RE-MODEL (TKR) | 37.7 | 40.5 | .003* | 36.4 | .017* |
| Major Bleeding (%) | |||||
| RE-NOVATE (THR) | 1.6 | 1.3 | 2.0 | ||
| RE-MOBILIZE (TKR) | 1.4 | 0.6 | 0.6 | ||
| RE-MODEL (TKR) | 1.3 | 1.3 | 1.5 | ||
| THR = total hip replacement; TKR = total knee replacement *Noninferior to enoxaparin. †Inferior to enoxaparin. |
|||||
In the phase 3 RE-LY trial, 2 different dosages of dabigatran etexilate (110 mg or 150 mg twice daily) were compared with dose-adjusted warfarin for stroke prevention in patients with atrial fibrillation and at least 1 risk factor for stroke.[34] The primary endpoint was the incidence of stroke or systemic embolism. RE-LY demonstrated that dabigatran etexilate 110 mg twice daily not only provided antithrombotic protection similar to well-managed warfarin but also was associated with a lower annual rate of major bleeding (3.36% vs 2.71%; P = .003). The twice-daily 150-mg dosage of dabigatran etexilate resulted in a lower rate of stroke/systemic embolism, 1.11% vs 1.69% (relative risk [RR], 0.66; 95% confidence interval [CI], 0.53-0.82; P < .001 for superiority) and was associated with a similar risk for major bleeding. Both dabigatran dose regimens were associated with significantly lower rates of intracranial hemorrhage than warfarin.
The efficacy and safety of dabigatran etexilate for the treatment of VTE was evaluated in RE-COVER.[35] This randomized, double-blind, noninferiority trial in patients with acute VTE who were initially given parenteral anticoagulation therapy for a median of 9 days compared dabigatran etexilate (150 mg twice daily) with warfarin (target INR = 2-3). The primary outcome was the 6-month incidence of recurrent symptomatic, objectively confirmed VTE and related deaths. The incidence of recurrent VTE was 2.4% in the dabigatran etexilate group vs 2.1% in the warfarin group, a difference that fell within the prespecified margin for noninferiority (P < .0001). Major bleeding occurred in 1.6% of dabigatran etexilate-treated patients and 1.9% of warfarin-treated patients. There were no significant differences in the number of deaths, acute coronary syndromes (ACS), and abnormal liver-function tests between the 2 treatment groups.
A second study with an identical design, RE-COVER II,[36] is underway as are the RE-MEDY[37] and RE-SONATE[38] trials, which are evaluating dabigatran etexilate for secondary prevention of VTE.
RE-DEEM, a multicenter, placebo-controlled, dose-finding, phase 2 study in patients with ACS, evaluated the safety and efficacy of dabigatran etexilate at 50 mg, 75 mg, 110 mg, and 150 mg twice daily vs placebo, administered within a few weeks of the acute event, in addition to dual antiplatelet therapy, over a 6-month period. The primary endpoint was the composite of major and clinically relevant minor bleeding. Secondary endpoints included death, MI, severe recurrent ischemia, stroke, and changes in D-dimer levels.[39] Results of RE-DEEM were presented at the American Heart Association 2009 Scientific Sessions. Dabigatran etexilate was associated with a dose-related increased risk for major or minor bleeding (the primary endpoint) vs placebo; although, the absolute increase in major bleeds was not more than 1%, according to study investigators (Table 3).[40]
Table 3. Bleeding and Cardiovascular Outcomes From the RE-DEEM Trial
| Placebo n = 371 |
Dabigatran Dose | ||||
| 50 mg BID | 75 mg BID n = 368 |
110 mg BID n = 406 |
150 mg BID n = 347 |
||
| 1O Major and clinically relevant minor bleeding |
2.4 | 3.5 | 4.3 | 7.9 | 7.8 |
| Major bleeding* | 0.5 | 0.8 | 0.3 | 2.0 | 1.2 |
| 2O: CV death, nonfatal MI, stroke |
3.8 | 4.6 | 4.9 | 3.0 | 3.5 |
| *International Society of Thrombosis and Hemostasis criteria | |||||
Rivaroxaban
Rivaroxaban is a direct FXa inhibitor that selectively and reversibly inhibits both free and clot-associated FXa activity, as well as prothrombinase activity.[41] Pharmacodynamic features of rivaroxaban are listed in Table 2.
The efficacy and safety of rivaroxaban in preventing VTE following major orthopaedic surgery was demonstrated in 4 phase 3 studies, RECORD1-4.[42-45] The primary efficacy endpoint, in all 4 studies, was the composite of DVT, nonfatal PE, and all-cause mortality (total VTE); the main secondary efficacy endpoint was the composite of proximal DVT, nonfatal PE, and VTE-related death (major VTE). The primary safety endpoint was major bleeding. RECORD1 and RECORD3 compared rivaroxaban 10 mg once daily (starting 6-8 hours after surgery) with enoxaparin 40 mg once daily (starting the evening before surgery) given for 31-39 days (extended prophylaxis) after THR (RECORD1)[42] and 10-14 days (short-term prophylaxis) after TKR (RECORD3).[44] RECORD2 investigated the efficacy and safety of extended (5 weeks; 31-39 days) thromboprophylaxis with rivaroxaban compared with short-term (10-14 days followed by placebo) enoxaparin in patients undergoing THR.[43] RECORD4 compared the efficacy and safety of rivaroxaban 10 mg once daily (starting 6-8 hours after surgery) with enoxaparin 30 mg twice daily (starting 12-24 hours after surgery), given 10-14 days in patients undergoing TKR.[45] In all 4 studies, rivaroxaban was superior to enoxaparin for the primary efficacy endpoint, with no significant difference in the rates of major or clinically relevant nonmajor bleeding between the treatment groups (Table 4).
Table 4. Efficacy and Safety Outcomes From the RECORD Studies
| Endpoint | RECORD1 (THR) | RECORD2 (THR) | RECORD3 (THR) | RECORD4 (THR) | ||||
| Enox 40 mg QD | Rivarox 10 mg QD | Enox 40 mg QD | Rivarox 10 mg QD | Enox 40 mg QD | Rivarox 10 mg QD | Enox 30 mg BID | Rivarox 10 mg QD | |
| 5 weeks | 5 weeks | 10−14 days |
5 weeks | 10−14 days |
10−14 days |
10−14 days |
10−14 days |
|
| Efficacy endpoints | ||||||||
| Total VTE % | 3.7 | 1.1 P<.001 |
9.3 | 2.0 P<.0001 |
18.9 | 9.6 P<.001 |
10.1 | 6.9 P=.012 |
| Major VTE % | 2.0 | 0.2 P<.001 |
5.1 | 0.6 P<.0001 |
2.6 | 1.0 P=.01 |
2.0 | 1.2 P=.124 |
| Sympto- matic VTE % |
0.5 | 0.3 P=.22 |
1.2 | 0.2 P=.004 |
2.0 | 0.7 P=.005 |
1.2 | 0.7 P=.187 |
| Bleeding endpoints | ||||||||
| Major bleeding % | 0.1 | 0.3 | < 0.1 | < 0.1 | 0.5 | 0.6 | 0.3 | 0.7 |
| Clinically relevant nonmajor bleeding % | 2.4 | 2.9 | 2.7 | 3.3 | 2.3 | 2.7 | 2.0 | 2.6 |
| All P values for efficacy calculated from absolute risk reduction | ||||||||
Rivaroxaban is undergoing evaluation for the treatment of DVT (EINSTEIN-DVT)[46] and PE (EINSTEIN-PE),[47] secondary prevention of VTE (EINSTEIN-EXT),[48] prevention of VTE in hospitalized medically ill patients, and (MAGELLAN),[49] stroke prevention in patients with AF (ROCKET-AF),[50] and secondary prevention of cardiovascular events in patients with ACS (ATLAS ACS-TIMI 51)[51] in large phase 3 trials. The results of the EINSTEIN-EXT were presented at the American Society of Hematology 2009 Meeting.[48]
EINSTEIN-EXT compared rivaroxaban 20 mg once daily vs placebo for an additional 6 or 12 months in patients who had completed 6-12 months of anticoagulant treatment with a VKA for acute VTE. Patients who had a clear indication for continued anticoagulant treatment were excluded from the study. Symptomatic recurrent VTE (composite of recurrent DVT, nonfatal PE, and fatal PE) was the primary efficacy endpoint. The primary and secondary safety endpoints were major bleeding and clinically relevant nonmajor bleeding, respectively. After a mean of 190 days, rivaroxaban-treated patients had a significant 82% relative risk reduction of symptomatic recurrent VTE events compared with placebo-treated patients (1.3% vs 7.1%; P < .0001). Bleeding events, however, were numerically greater among patients treated with rivaroxaban. Major bleeding occurred in 0.7% of rivaroxaban recipients compared with 0% in placebo recipients (P = .106) although none of the major bleeding events was fatal or in a critical site. Clinically relevant nonmajor bleeding was reported in 5.4% and 1.2% of the rivaroxaban and placebo arms, respectively.[48]
The ATLAS ACS-TIMI 46 study assessed the safety and efficacy of rivaroxaban in patients who had been stabilized after an ACS event.[52] In this double-blind, dose-escalation, phase 2 study, patients were initially stratified into 2 groups: those who were to receive aspirin only or those who were to receive aspirin plus a thienopyridine. Within each group, patients were then randomized to receive either placebo or rivaroxaban at doses of 5-20 mg, with the total dose given once daily or twice daily. The primary safety endpoint was clinically significant bleeding (TIMI major, TIMI minor, or bleeding requiring medical attention); the primary efficacy endpoint was death, MI, stroke, or severe recurrent ischemia requiring revascularization during 6 months. The main secondary efficacy endpoint was death, MI, or stroke.
Rivaroxaban was associated with a dose-dependent increase in clinically significant bleeding events compared with placebo (hazard ratio [HR], 2.21; 95% CI, 1.25-3.91 for 5 mg; HR, 3.35; 95% CI: 2.31-4.87 for 10 mg; HR, 3.60; 95% CI, 2.32-5.58 for 15 mg; and HR, 5.06; 95% CI, 3.45-7.42 for 20 mg doses; P < .0001. Although there was a trend toward a reduction in the primary efficacy endpoint vs placebo, this did not reach statistical significance (5.6% vs 7.0%; HR, 0.79; 95% CI, 0.60-1.05; P = .10). However, rivaroxaban reduced the main secondary efficacy endpoint compared with placebo (3.9% vs 5.5%; HR, 0.69; 95% CI, 0.50-0.96; P = .027). Based on the results from this study, the phase 3 study ATLAS ACS-TIMI 51 is underway to evaluate the efficacy of low-dose rivaroxaban in reducing major ischemic outcomes in ACS patients.[51]
Apixaban
Apixaban, like rivaroxaban, is a direct FXa inhibitor that selectively and reversibly inhibits both free and clot-associated FXa activity, as well as prothrombinase activity[53] (Table 2).
Three phase 3 trials, ADVANCE-1, ADVANCE-2, and ADVANCE-3, have evaluated the efficacy and safety of apixaban for the prevention of VTE after major orthopaedic surgery. The primary efficacy outcome of these studies was the composite of DVT, PE, and death from any cause during the treatment period. In ADVANCE-1, apixaban 2.5 mg twice daily did not meet the criteria for noninferiority compared with enoxaparin 30 mg twice daily for prevention of VTE in patients undergoing TKR, but the rate of major bleeding was lower with apixaban[54] (Table 5). In ADVANCE-2, the noninferiority of apixaban to enoxaparin (40 mg once daily) was demonstrated in patients undergoing TKR.[55] In both trials, bleeding rates were lower with apixaban. Results of the recently completed ADVANCE-3 study that assessed extended prophylaxis with apixaban compared with enoxaparin 40 mg once daily (35 days for both regimens) in patients undergoing THR are awaited.[56]
Table 5: Efficacy and Safety Outcomes From ADVANCE-1 and ADVANCE-2
| ADVANCE-1 | ADVANCE-2 | |||
| Enoxaparin 30 mg BID | Apixaban 2.5 mg BID | Enoxaparin 40 mg QD | Apixaban 2.5 mg BID | |
| 10-14 days | 10-14 days | 10-14 days | 10-14 days | |
| DVT, PE, and all-cause mortality (%) |
8.8 | 9.0 P = .06† |
24.37 | 15.06 P < .0001* |
| Major bleeding (%) | 0.7 | 1.4 P = .053 |
0.9 | 0.6 P = .3014 |
| †Inferior to enoxaparin *Noninferior to enoxaparin |
||||
Further large phase 3 studies with apixaban are in progress, evaluating its efficacy and safety for primary prophylaxis of VTE in hospitalized medically ill patients (ADOPT),[57] secondary prevention of VTE (AMPLIFY-EXT),[58] stroke prevention in AF (ARISTOTLE),[59] and secondary prevention of cardiovascular events in ACS (APPRAISE-2).[60]
Encouraging results from the APPRAISE study showing a trend toward a reduction in ischemic events with the addition of apixaban to antiplatelet therapy in patients with recent ACS formed the basis for the ongoing APPRAISE-2 trial. APPRAISE was a phase 2, double-blind, placebo-controlled, dose-ranging study in patients with recent ST-elevation or non-ST-elevation ACS.[61] Patients were randomized to 6 months of placebo or 1 of 4 doses of apixaban: 2.5 mg twice daily, 10 mg once daily, 10 mg twice daily, or 20 mg once daily. All patients also received background antiplatelet therapy (~ 100% aspirin and 76% clopidogrel). The primary outcome in this study was the rate of bleeding, which was defined using the International Society on Thrombosis and Haemostasis definitions of major or clinically relevant nonmajor bleeding. Cardiovascular death, MI, severe recurrent ischemia, or ischemic stroke was a secondary outcome. Prior to the completion of the study the 10 mg twice daily and the 20 mg once daily doses of apixaban were discontinued because of excess total bleeding. The other 2 doses were associated with a dose-related increase in major or clinically relevant nonmajor bleeding compared with placebo: apixaban 2.5 mg twice daily (HR, 1.78; 95% CI, 0.91-3.48; P = .09) and 10 mg once daily (HR, 2.45; 95% CI, 1.31-4.61; P = .005). However, the lower doses produced a reduction in ischemic events compared with placebo: apixaban 2.5 mg twice daily (HR, 0.73; 95% CI, 0.44-1.19; P = .21) and 10 mg once daily (HR, 0.61; 95% CI, 0.35-1.04; P = .07).
Emerging Targets for New Anticoagulants
The currently available anticoagulant agents all target thrombin or FXa, either indirectly or directly. Thrombin is a logical target because of its multiple roles in coagulation. It converts fibrinogen to fibrin, the final step in the coagulation cascade; it amplifies its own generation by feedback activation of FV and FVIII, key cofactors for the prothrombinase and intrinsic tenase complex, respectively; and it coordinates platelet activation and aggregation with coagulation. The inhibition of thrombin not only blocks fibrin formation but also attenuates thrombin generation and platelet activation.[62] FXa also has a central role in the process of coagulation and platelet activation. FXa is an essential component of the prothrombinase complex that triggers the generation of thrombin. Thus, the inhibition of FXa and subsequent thrombin generation represent an important strategy for new anticoagulant drugs.[63]
Given that the sequential activation of coagulation factors results in an amplification at each step, an anticoagulant that targets an upstream factor, such as TF-FVIIa complex, might be more potent than an agent that targets downstream factors, like thrombin and FXa.[6] The caveat, however, is that TF-FVIIa complex is essential for hemostasis and inhibition of this complex may result in extensive bleeding.[64] In mice, gene-knockout studies have shown that TF, as well as FVII, FX, FV, and prothrombin (factors involved in the initiation and propagation phases of coagulation) are essential for hemostasis and for survival.[65] Nonetheless, NAPc2, a compound isolated from the canine hookworm, which inhibits the TF-FVIIa complex by preventing it from binding to FX, successfully completed phase 2 trials.[66] Factors involved in the amplification of coagulation (FVIII, FIX, FXI, and FXII), in contrast, are not essential, for mice lacking these factors are able to survive.[65] Further deficiency, in mice, of FXII or FXI was shown to attenuate the development of thrombosis but was not associated with impaired hemostasis.[67] Intrinsic pathway factors may thus represent potential targets for therapy as their inhibition might mitigate bleeding risks. However, in the setting of ongoing thrombosis, inhibition of the intrinsic pathway components may have less impact than would inhibition of downstream components such as FX and thrombin.[6]
Conclusions
The coagulation cascade is responsible for maintaining vascular integrity and inducing rapid clot formation after vessel injury. Under pathological conditions, excessive quantities of thrombin can form, triggering thrombosis. Thrombin and FXa remain the main targets of traditional and the new anticoagulants.
The new oral anticoagulants have numerous advantages over warfarin; most importantly, they can be given in fixed doses without anticoagulation monitoring, thereby streamlining therapy. With promising results of phase 2 and 3 trials, these new agents are likely to slowly replace warfarin for many indications. Whether agents that target upstream coagulation factors such as FXIIa or FXIa will be as effective as these new agents, requires further evaluation.
Supported by an independent educational grant from Bayer Healthcare.
This article is a CME certified activity. To earn credit for this activity visit:
http://cme.medscape.com/viewarticle/725813
Trial names used in this text
| ADOPT | Apixaban for the Prevention of Thrombosis-related Events in Patients With Acute Medical Illness. http://www.clinicaltrials.gov/ct2/show/NCT00457002 |
| ADVANCE | An Investigational Drug for the Prevention of Thrombosis-related Events Following Knee Replacement Surgery. http://www.clinicaltrials.gov/ct2/show/NCT00452530 |
| AMPLIFY-EXT | Efficacy and Safety Study of Apixaban for Extended Treatment of Deep Vein Thrombosis or Pulmonary Embolism. http://clinicaltrials.gov/ct2/show/NCT00633893 |
| APPRAISE | Apixaban for Prevention of Acute Ischemic Events: A Phase 3, Randomized, Double-Blind, Evaluation of the Safety and Efficacy of Apixaban in Subjects with a Recent Acute Coronary Syndrome. http://www.clinicaltrials.gov/ct2/show/NCT00831441 |
| ARISTOTLE | Apixaban for the Prevention of Stroke in Subjects With Atrial Fibrillation. http://www.clinicaltrials.gov/ct2/show/NCT00412984 |
| ATLAS ACS-TIMI 51 | Rivaroxaban in Combination With Aspirin Alone or With Aspirin and a Thienopyridine in Subjects With Acute Coronary Syndromes. Thrombolysis in Myocardial Infarction 51. http://clinicaltrials.gov/ct2/show/NCT00809965 |
| ATLAS-TIMI 46 | Anti-Xa Therapy to Lower cardiovascular events in addition to Aspirin with or without thienopyridine therapy in Subjects with Acute Coronary Syndrome-Thrombolysis In Myocardial Infarction 46. http://clinicaltrials.gov/ct2/show/NCT00402597 |
| EINSTEIN-DVT | Oral Direct Factor Xa Inhibitor Rivaroxaban in Patients with Acute Symptomatic Deep-vein Thrombosis Without Symptomatic Pulmonary Embolism. http://clinicaltrials.gov/ct2/show/NCT00440193 |
| EINSTEIN-EXT | Once – Daily Oral Direct Factor Xa Inhibitor Rivaroxaban In The Long-Term Prevention Of Recurrent Symptomatic Venous Thromboembolism In Patients With Symptomatic Deep-Vein Thrombosis Or Pulmonary Embolism. http://www.clinicaltrials.gov/ct2/show/NCT00439725 |
| EINSTEIN-PE | Oral Direct Factor Xa Inhibitor Rivaroxaban in Patients with Acute Symptomatic Pulmonary Embolism with or without Symptomatic Deep-vein Thrombosis. http://clinicaltrials.gov/ct2/show/NCT00439777 |
| MAGELLAN | Venous Thromboembolic Event (VTE) Prophylaxis in Medically ill Patients. http://clinicaltrials.gov/ct2/show/NCT00571649 |
| RE-COVER | Dabigatran versus Warfarin in the Treatment of Acute Venous Thromboembolism. http://clinicaltrials.gov/show/NCT00291330 |
| RE-DEEM | Dose Finding Study for Dabigatran Etexilate in Patients With Acute Coronary Syndrome. http://www.clinicaltrials.gov/ct2/show/NCT00621855 |
| RE-LY | Randomized Evaluation of Long Term Anticoagulant Therapy. http://www.clinicaltrials.gov/ct2/show/NCT00262600 |
| RE-MEDY | A Randomised, Multicenter, Double-blind, Active Controlled Study to Investigate the Efficacy and Safety of Dabigatran Etexilate Compared to Warfarin for the Secondary Prevention of Venous Thromboembolism. http://clinicaltrials.gov/ct2/show/NCT00329238 |
| RE-MOBILIZE | Dabigatran Etexilate vs Enoxaparin in Prevention of VTE Post Total Knee Replacement. http://www.clinicaltrials.gov/ct2/show/NCT00152971 |
| RE-MODEL | Thromboembolism prevention after knee surgery. http://www.clinicaltrials.gov/ct2/show/NCT00168805 |
| RE-NOVATE | Dabigatran Etexilate Compared with Enoxaparin in Prevention of VTE Following Total Hip Arthroplasty. http://www.clinicaltrials.gov/ct2/show/NCT00657150 |
| RE-SONATE | Twice-daily oral direct thrombin inhibitor dabigatran etexilate in the long-term prevention of recurrent symptomatic proximal venous thromboembolism in patients with symptomatic deep-vein thrombosis or pulmonary embolism. http://clinicaltrials.gov/ct2/show/NCT00558259 |
| RECORD | Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Venous Thrombosis and Pulmonary Embolism. RECORD1: http://www.clinicaltrials.gov/ct2/show/NCT00329628 RECORD2: http://www.clinicaltrials.gov/ct2/show/NCT00332020 RECORD3: http://www.clinicaltrials.gov/ct2/show/NCT00361894 RECORD4: http://www.clinicaltrials.gov/ct2/show/NCT00362232 |
| ROCKET-AF | Randomized, Double-blind study comparing once daily oral rivaroxaban with adjusted-dose oral warfarin for the prevention of stroke in subjects with non-valvular atrial fibrillation. http://clinicaltrials.gov/ct2/show/NCT00403767 |
References
- Macfarlane RG. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature. 1964;202:498-499.
- Davie EW, Ratnoff OD. Waterfall sequence for intrinsic blood clotting. Science. 1964;145:1310-1312.
- Hoffman M, Monroe DM. A cell-based model of hemostasis. Thromb Haemost. 2001;85:958-965.
- Hoffman M, Monroe DM. Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007;21:1-11.
- Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938-949.
- Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451:914-918.
- Ignarro LJ, Buga GM, Wood KS, et al. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265-9269.
- Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524-526.
- Marcus AJ, Broekman MJ, Pinsky DJ. COX inhibitors and thromboregulation. N Engl J Med. 2002;347:1025-1026.
- Marcus AJ, Broekman MJ, Drosopoulos JH, et al. Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin Thromb Hemost. 2005;31:234-246.
- Semeraro N, Colucci M. Tissue factor in health and disease. Thromb Hemost. 1997;78:759-764.
- Grignani G, Maiolo A. Cytokines and hemostasis. Haematologica. 2000;85:967-972.
- Panes O, Matus V, Sáez CG, Quiroga T, Pereira J, Mezzano D. Human platelets synthesize and express functional tissue factor. Blood. 2007;109:5242-5250.
- Rao LV, Pendurthi UR. Tissue factor-factor VIIa signalling. Arterioscler Thromb Vasc Biol. 2005;25:47-56.
- Mann KG, Brummel-Ziedins K, Orfeo T, Butenas S. Models of blood coagulation. Blood Cells Mol Dis. 2006;36:108-117.
- Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;12:17-25.
- Bouma BN, Mosnier LO. Thrombin activatable fibrinolysis inhibitor (TAFI) at the interface between coagulation and fibrinolysis. Pathophysiol Haemost Thromb. 2004;33:375-381.
- Bronze GJ. Tissue factor pathway inhibitor. Thromb Haemost. 1995;74:90-93.
- Pike RN, Buckle AM, le Bonnie BF, et al. Control of the coagulation system by serpins; getting by with a little help from glycosaminoglycans. FEBS J. 2005;272:4842-4851.
- Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006;32:49-60.
- Corrál J, González-Conejero R, Hernandez-Espinosa D, et al. Protein Z/Z-dependent protease inhibitor (PZ/ZPI) anticoagulant system and thrombosis. Br J Haematol. 2007;137:99-108.
- Hoffman R, Benz EJ, Shattil SJ, Furie B, Silberstein LE (eds). Hematology: Basic Principles and Practice, 5th ed. Philadelphia, PA:Elsevier, 2008.
- Geerts W, Bergquist D, Pineo F, et al. Prevention of venous thromboembolism. American College of Chest Physicians’ evidence-based clinical practice guidelines (8th ed). Chest. 2008;133:381S-453S.
- Suttie JW. Mechanism of action of vitamin K: Synthesis of gamma-carboxyglutamic acid. CRC Crit Rev Biochem. 1980;8:191-223.
- Bounameaux H, Perrier A. Duration of anticoagulation therapy for venous thromboembolism. Hematology Am Soc Hematol Educ Program. 2008:252-258.
- Ansell J, Hirsh J, Poller L, et al. The pharmacology and management of the vitamin K antagonists: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest. 2004;126:204-233.
- Hirsh J, O’Donnell M, Eikelboon JW. Beyond unfractionated heparin and warfarin: current and future advances. Circulation. 2007;116:552-560.
- Bond CA, Raehl CL. Pharmacist-provided anticoagulation management in United States hospitals: death rates, length of stay, medicare charges, bleeding complications, and transfusions. Pharmacotherapy. 2004;24:953-963.
- Gross PL, Weitz JI. New antithrombotic drugs. Clin Pharmacol Ther. 2009;86:139-146.
- Stangier J, Rathgen K, Stahle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol. 2007;64:292-303.
- Eriksson BI, Dahl OE, Rosencher N, , et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: A randomised, double-blind, non-inferiority trial. Lancet. 2007;370:949-956.
- The RE-MOBILIZE Writing Committee. The oral thrombin inhibitor dabigatran etexilate vs the North American enoxaparin regimen for the prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty. 2009;24:1-9.
- Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: The Re-model randomized trial. J Thromb Haemost. 2007;5:2178-2185.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361:2342-2352.
- RE-COVER II. A phase III, randomised, double blind, parallel-group study of the efficacy and safety of oral dabigatran etexilate (150 mg bid) compared to warfarin (INR 2.0-3.0) for 6 month treatment of acute symptomatic venous thromboembolism, following initial treatment (5-10 days) with a parenteral anticoagulant approved for this indication.” Available at: http://www.clinicaltrials.gov/ct2/show/NCT00680186 Accessed June 5, 2010.
- RE-MEDY. A randomised, multicenter, double-blind, active controlled study to investigate the efficacy and safety of dabigatran etexilate, 150 mg b.i.d administered orally (capsules) for 18 months, compared to warfarin tablets p.r.n. (target INR) for the secondary prevention of venous thromboembolism. Available at: http://clinicaltrials.gov/ct2/show/NCT00329238 Accessed June 5, 2010.
- RE-SONATE. Twice-daily oral direct thrombin inhibitor dabigatran etexilate in the long-term prevention of recurrent symptomatic proximal venous thromboembolism in patients with symptomatic deep-vein thrombosis or pulmonary embolism. Available at: http://clinicaltrials.gov/ct2/show/NCT00558259 Accessed June 5, 2010.
- Oldgren J, Budaj A, Granger CB, et al. Randomised dabigatran etexilate dose finding study in patients with acute coronary syndromes post index event with additional risk factors for cardiovascular complications also receiving aspirin and clopidogrel (RE-DEEM). Circulation. 2009;120;2162.
- Oldgren J. Randomised dabigatran etexilate dose finding study in patients with acute coronary syndromes post index event with additional risk factors for cardiovascular complications also receiving aspirin and clopidogrel (RE-DEEM). American Heart Association 2009 Scientific Sessions; November 18, 2009b; Orlando, FL. Available at: http://www.theheart.org/article/1026791.do Accessed June 5, 2010.
- Perzborn E, Strassburger J, Wilmen A, et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939 – an oral, direct Factor Xa inhibitor. J Thromb Haemost. 2005;3:514-521.
- Eriksson BI, Borris LC, Friedman RJ, et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med. 2008;358:2765-2775.
- Kakkar AK, Brenner B, Dahl OE, et al. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomized controlled trial. Lancet. 2008;372:31-39.
- Lassen MR, Ageno W, Borris LC, et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med. 2008;358:2776-2786.
- Turpie AG, Lassen MR, Davidson BL, et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial. Lancet. 2009;373:1673-1680.
- EINSTEIN-DVT. Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic deep-vein thrombosis without symptomatic pulmonary embolism: Einstein-DVT evaluation. Available at:http://clinicaltrials.gov/ct2/show/NCT00440193 Accessed June 5, 2010.
- EINSTEIN-PE. Oral direct factor Xa inhibitor rivaroxaban in patients with acute symptomatic pulmonary embolism with or without symptomatic deep-vein thrombosis: EINSTEIN-PE evaluation. Available at:http://clinicaltrials.gov/ct2/show/NCT00439777 Accessed June 5, 2010.
- Buller HR. Once-daily oral rivaroxaban versus placebo in the long-term prevention of recurrent symptomatic venous thromboembolism. The Einstein-Extension Study. The American Society of Hematology meeting 2009 Abstract. Available at: http://ash.confex.com/ash/2009/webprogram/Paper25669.html Accessed June 5, 2010.
- MAGELLAN. Venous thromboembolic event (VTE) prophylaxis in medically ill patients (MAGELLAN). Available at: http://clinicaltrials.gov/ct2/show/NCT00571649 Accessed June 5, 2010.
- ROCKET-AF. Randomized, double-blind study comparing once daily oral rivaroxaban with adjusted-dose oral warfarin for the prevention of stroke in subjects with non-valvular atrial fibrillation. Available at:http://clinicaltrials.gov/ct2/show/NCT00403767 Accessed June 5, 2010.
- ATLAS ACS TIMI 51. An efficacy and safety study for rivaroxaban in patients with acute coronary syndrome. Available at: http://clinicaltrials.gov/ct2/show/NCT00809965 Accessed June 5, 2010.
- Mega JL, Braunwald E, Mohanavelu S, et al; ATLAS ACS-TIMI 46 study group. Rivaroxaban versus placebo in patients with acute coronary syndromes (ATLAS ACS-TIMI 46): a randomised, double-blind, phase II trial. Lancet. 2009;374:29-38.
- Harenberg J, Wehling M. Current and future prospects for anticoagulant therapy: inhibitors of factor Xa and factor IIa. Semin Thromb Hemost. 2008;34:39-57.
- Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med. 2009;361:594-604.
- Lassen MR, Raskob GE, Gallus A, et al; ADVANCE-2 investigators. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet. 2010;375:807-815.
- ADVANCE-3. Study of an investigational drug for the prevention of thrombosis-related events following hip replacement surgery. Available at: http://clinicaltrials.gov/ct2/show/NCT00423319 Accessed June 5, 2010.
- ADOPT. Study of apixaban for the prevention of thrombosis-related events in patients with acute medical illness (ADOPT). Available at: http://clinicaltrials.gov/ct2/show/NCT00457002 Accessed June 5, 2010.
- AMPLIFY-EXT. A safety and efficacy trial evaluating the use of apixaban for the extended treatment of deep vein thrombosis and pulmonary embolism. Available at: http://clinicaltrials.gov/ct2/show/NCT00633893 Accessed June 5, 2010.
- ARISTOTLE. Apixaban for the prevention of stroke in subjects with atrial fibrillation (ARISTOTLE). Available at:http://clinicaltrials.gov/ct2/show/NCT00412984 Accessed June 5, 2010.
- APPRAISE-2. Phase III acute coronary syndrome (APPRAISE-2). Available at: http://clinicaltrials.gov/ct2/show/NCT00831441 Accessed June 5, 2010.
- APPRAISE Steering Committee and Investigators. Apixaban, an oral, direct, selective factor Xa inhibitor, in combination with antiplatelet therapy after acute coronary syndrome: results of the Apixaban for Prevention of Acute Ischemic and Safety Events (APPRAISE) trial. Circulation. 2009;119:2877-2885.
- Weitz JI. Factor Xa or thrombin: is thrombin a better target? J Thromb Haemost. 2007;5:65-67.
- Ansell J. Factor Xa or thrombin: is factor Xa a better target? J Thromb Haemost. 2007;5:60-64.
- Snyder LA, Rudnick KA, Tawadros R, et al. Expression of human tissue factor under the control of the mouse tissue factor promoter mediates normal hemostasis in knock-in mice. J Thromb Haemost. 2008;6:306-314.
- Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25:2273-2281.
- Giuliano RP, Wiviott SD, Stone PH, et al. Recombinant nematode anticoagulant protein c2 in patients with non-ST-segment elevation acute coronary syndrome: The ANTHEM-TIMI-32 trial. J Am Coll Cardiol. 2007;49:2398-2407.
- Galiani D, Renne T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost. 2007;5:1106-1112.
Tags: Rianimazione




One Response to “Coagulation Simplified”
Trackbacks/Pingbacks
Leave a Reply
You must be logged in to post a comment.